1.本发明属于生物医用高分子材料技术领域,涉及一种功能化聚碳酸酯及其制备方法,可用作血液接触制品。
背景技术:
2.随着医学技术的进一步发展,对医疗器械的需求也进一步提高。应用在特殊领域的医疗器械,要求材料具有一些特殊性能。在血液治疗过程中如体外的血液透析、血液灌流,体内的血管支架移植,要求材料具有良好的血液相容性
3.聚碳酸酯(pc)作为一种工程塑料,由于其良好的机械性能、耐热性能、光学性能及耐化学性能被广泛的应用于汽车、电子设备,建筑、办公用品、光盘、运动器材、医疗保健、计算机、航空航天等领域。然而,聚碳酸酯在血液接触材料领域的应用较少,由于其血液相容性较差,与血液直接接触会引起血小板聚集激活进而形成血栓以及破坏血细胞等一系列不良影响。
4.有研究指出利用与具有生物活性的物质共混的方式来改善聚碳酸酯的生物相容性。例如专利cn105062029b中将聚碳酸酯和甲壳素、羟丙基甲基纤维素共混,制备具有聚碳酸酯生物净化材料。专利cn108815590a将玉米淀粉、阿拉伯胶、甘露聚糖、丝素蛋白等组分与聚乙烯碳酸酯、聚碳酸酯共混,制备多糖-丝素蛋白复合抗凝血生物材料。但是利用共混的方法提高聚碳酸酯的生物相容性,通常会加入小分子的增塑剂提高各组分间的相容性。存在小分子增塑剂迁移、泄露的风险,这会对人体造成不良的影响。此外通过与纤维素、淀粉、多糖、蛋白等天然大分子共混的方式提高聚碳酸酯的生物相容性,会对聚碳酸酯本身的性能造成不良影响,如模量降低、冲击强度变差、热学性能降低等。
5.通过共聚的方法提高聚碳酸酯的血液相容性是一种更安全有效的方法。cn113402704a中提供了一种制备羧基功能化的聚碳酸酯的制备方法,改善了聚碳酸酯的血液相容性,使其与血液接触时不会造成不良的影响。但是由于其不具备自抗凝性,在制备成如血液透析膜或者血液灌流微球用于肾衰竭或肝衰竭的体外治疗时仍需要另外加入抗凝剂避免形成血栓。传统抗凝剂直接注射使用,因此会随着血液循环回到体内导致其在治疗结束后依然发挥抗凝血作用,使得病人存在出血倾向。而且,大部分抗凝剂需通过肾脏或者肝代谢,然而接受血液净化的患者大部分为肾衰竭或肝衰竭患者,肾滤过率或者肝代谢能力下降,体内的抗凝剂清除时间延长进一步加剧了出血倾向。此外,由于每个患者的体差异性临床医生往往很难确定抗凝剂准倾向。
技术实现要素:
6.针对现有技术中存在的上述问题,本发明的目的在于提供一种功能化的聚碳酸酯及其制备方法,其具有良好血液相容性,特别的具有自抗凝性、低的溶血率以及抑制血小板激活,同时保持聚碳酸酯原本的性能,具有良好的力学性能及可加工性,可用作血液接触制品,例如可以作为体外治疗肾衰竭及肝衰竭血液等的净化材料。
7.为达到以上发明目的,本发明的技术方案如下:
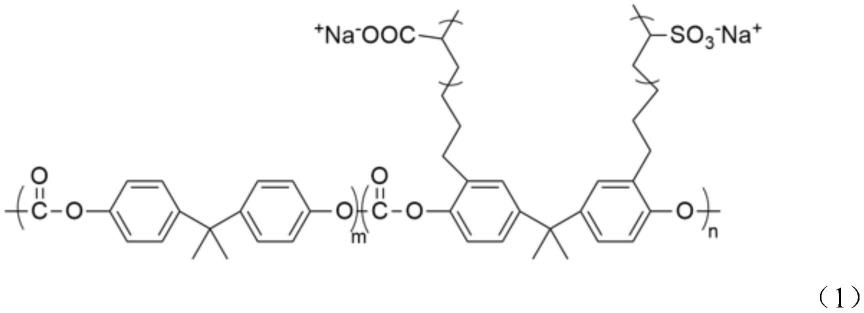
8.本发明提供一种功能化的聚碳酸酯,其分子侧链上同时包括含有羧基和磺酸基基团,具有如式(1)所示的结构:
[0009][0010]
式中,m取值为1-10,优选4-7;n取值为1-10,优选3-6。
[0011]
本发明所述功能化的聚碳酸酯,其重均分子量为23000-36000g/mol,优选25000-28000g/mol。
[0012]
本发明还提供一种上述功能化聚碳酸酯的制备方法,该方法分为两步进行,涉及自由基及缩聚两种聚合方法,具有反应条件温和、能量消耗少、产品质量稳定。
[0013]
本发明所述功能化聚碳酸酯的制备方法,具体包括以下步骤:
[0014]
(1)将丙烯酸钠、对苯乙烯磺酸钠、二烯丙基双酚化合物、引发剂、水混合,氮气保护下在60-80℃,优选70-75℃预聚合反应2-24h,优选6-12h,得到预聚物;
[0015]
(2)氮气环境中,将步骤(1)的预聚物、双酚型化合物、碱金属氢氧化物、封端剂、水混合,搅拌得到澄清溶液,然后加入惰性有机溶剂,搅拌得到水油混合体系;
[0016]
(3)氮气保护下,将光气通入到步骤(2)的水油混合体系中,同时加入催化剂,在25-35℃,优选30-32℃下聚合反应0.5-4h,优选2-3h,得到功能化聚碳酸酯。
[0017]
本发明步骤(1)中,所述二烯丙基双酚化合物选自二烯丙基联苯二酚、二烯丙基双酚a、二烯丙基双酚z、二烯丙基双酚f、二烯丙基双酚s等中的至少一种,优选二烯丙基双酚a;
[0018]
所述引发剂选自过硫酸铵、过硫酸钾、过硫酸钠中的至少一种,优选过硫酸铵。
[0019]
本发明步骤(1)中,所述丙烯酸钠与对苯乙烯磺酸钠的摩尔比为1:(1-5),优选1:(3-3.5);
[0020]
所述二烯丙基双酚化合物用量为丙烯酸钠与对苯乙烯磺酸钠总摩尔量的0.5-1.5%,优选1-1.2%;
[0021]
所述引发剂的用量为丙烯酸钠与对苯乙烯磺酸钠总摩尔量的0.05-1%,优选0.1-0.5%。
[0022]
本发明步骤(1)中,所述水为超纯水;
[0023]
所述水的用量与丙烯酸钠和对苯乙烯磺酸钠总质量的比例为(12.5-20):1,优选(16-16.5):1。
[0024]
本发明步骤(1)中,所述预聚物,其重均分子量为1000-10000g/mol,优选3000-5000g/mol。
[0025]
本发明步骤(2)中,所述双酚型化合物选自双酚a、双酚b、双酚e、双酚f、双酚z、双酚tmc中的至少一种,优选双酚a;
[0026]
所述碱金属氢氧化物选自氢氧化钾、氢氧化钠、氢氧化锂、氢氧化铯中的至少一种,优选为氢氧化钠;
[0027]
所述封端剂选自苯酚、对甲基苯酚、对异丙基苯酚、对叔丁基苯酚中的至少一种,优选对叔丁基苯酚;
[0028]
所述惰性有机溶剂选自二氯甲烷、三氯甲烷、二氯乙烷、三氯乙烷中的至少一种,优选为二氯甲烷。
[0029]
本发明步骤(2)中,所述预聚物与双酚型化合物的质量比为1:(1.5-15.5),优选1:(3-4);
[0030]
所述封端剂为双酚型化合物质量的1-3%,优选1.5-2.5%;
[0031]
所述双酚型化合物与水的质量比为1:(12.5-20),优选1:(16-16.5);
[0032]
所述碱金属氢氧化物在澄清溶液中的质量浓度为2-8wt%,优选4-6wt%。
[0033]
所述水与惰性有机溶剂的质量比为1:(0.5-1.2),优选1:(1-1.05)。
[0034]
本发明步骤(3)中,所述光气通入到步骤(2)的水油混合体系中的用量,基于步骤(2)的水油混合体系中的预聚物与双酚型化合物(即共聚单体)总量计,预聚物和双酚型化合物总摩尔量与光气的摩尔量比例为1:(1-1.15),优选1:(1-1.12);
[0035]
所述光气采用连续进料方式,保持在反应过程中持续通入,光气通入时间与反应时间相同。
[0036]
本发明步骤(3)中,所述催化剂选自三乙胺、四丁基溴化铵、四丁基氯化铵中的至少一种,优选为三乙胺;
[0037]
所述催化剂的用量,基于步骤(2)的水油混合体系中的双酚型化合物计,催化剂与双酚型化合物的质量比为0.0001-0.005:1,优选为0.0002
ꢀ‑
0.0003:1。
[0038]
本发明步骤(3)中,所述聚合反应,反应体系的ph保持为11-12;可以通过加入氢氧化钠水溶液等常规手段进行调控,本发明不做具体限定。
[0039]
本发明步骤(3)中,所述聚合反应完成后还包括后处理过程,将聚合反应得到的聚碳酸酯共聚物乳液进行纯化后得到功能化聚碳酸酯产物;该纯化操作为本发明可以采用本领域常规方法,没有特别要求,例如可以采用专利cn202010666164.3、cn202010698811.9中所公开的后处理方法,具体如:共聚物乳液首先进行油水分离,之后依次进行洗涤、脱挥、粉碎、干燥得到粉料。
[0040]
本发明上述功能化聚碳酸酯以丙烯酸钠、对苯乙烯磺酸钠的预聚体和双酚型化合物作为单体,与光气通过光气界面缩聚法制备。制得的功能化聚碳酸酯具有自抗凝性、低的溶血率以及抑制血小板激活等性能,同时保持聚碳酸酯原本的性能,可以用作血液接触制品,例如可以作为固体抗凝剂用于血液净化材料,制备血液透析器、灌流器的外壳、血液透析膜、血液灌流吸附微球等。
[0041]
本发明所述功能化聚碳酸酯,表现为相较于贫血小板血浆有延长的活化部分凝血酶原时间,优选50-600s,更加优选为100-600s。
[0042]
本发明所述功能化聚碳酸酯,同时表现为具有延长的凝血酶时间,优选20-180s,更加优选的90-180s。
[0043]
本发明所述功能化聚碳酸酯,还具有良好的血细胞相容性,表现为低的溶血率,优选<5%,更优选<1%。与现有技术相比,本发明技术方案的有益效果在于:
[0044]
本发明通过共聚合的方式在侧链上同时引入特定结构的羧基和磺酸基基团,赋予聚碳酸酯良好的血液相容性,使其具有自抗凝性、低的溶血率及抑制血小板的激活,使用时不需要另外加入抗凝剂,减少病人出血以及并发症的风险,拓宽了聚碳酸酯的应用领域,为血液治疗领域的发展提供材料解决方案,在血液接触材料领域有广阔的应用前景。
[0045]
本发明所制备的功能化聚合物的分子量以及组成还可以进行调控,并且该方法步骤简单,便于实现工业化,有良好的工业化应用前景。
具体实施方式
[0046]
为了更好的理解本发明的技术方案,下面结合实施例进一步阐述本发明的内容,但本发明的内容并不仅仅局限于以下实施例。
[0047]
本发明实施例和对比例中采用的主要分析评价方法如下:
[0048]
分子量通过凝胶渗透色谱(gpc)法测试,采用型号为waters 1515的凝胶渗透色谱仪测试,流动相为二氯甲烷,温度为30℃。
[0049]
拉伸模量及屈服强度通过万能拉力机测试,采用型号为zwick,测试标准iso527。
[0050]
红外测试通过衰减全反射红外(atr-ftir)光谱仪测试,采用美国赛默飞(thermo fisher)公司的nicolet 560测试。
[0051]
核磁测试采用burker公司型号为av iii hd 400mhz的核磁共振波谱仪测试。
[0052]
体积熔融指数(mvr)通过熔指仪测试,测试条件为300℃,1.2kg。
[0053]
凝血反应测试利用半自动凝血仪(sysmex corporation,kobe,japan)测定活化部分凝血酶时间(aptt)和凝血酶时间(tt);
[0054]
具体的实验步骤如下:首先将1cm*1cm的pc薄膜在生理盐水中浸泡过夜并在37℃下孵化1h;之后去掉生理盐水,并在装有材料的孔板中加入300μl新鲜的贫血小板血浆(ppp),37℃下共孵化30min。对于aptt的测试,依次在测试杯中加入50μl孵化过的ppp、50μl aptt试剂、50μl 0.025m cacl2溶液。对于tt的测试,首先加入50μl孵化过的ppp到测试杯中,然后加入100μl凝血酶试剂。
[0055]
溶血率的测试按照astmf-756-08中规定的材料溶血率测试标准进行。
[0056]
血小板因子4(pf4)用来研究材料的血小板激活情况,pf4的浓度通过酶联免疫吸附(elisa)试剂盒测试;
[0057]
具体实验步骤如下:1cm*1cm的pc薄膜首先在生理盐水中浸泡过夜,并在37℃下孵化1h,之后将其与全血在37℃下共孵化2h。处理后的全血以1000rpm的转速离心15min,获得血浆用于后续的测试。后续的实验步骤根据各自的elisa试剂盒的具体要求进行操作。
[0058]
本发明各实施例和对比例中各原材料均为通过阿拉丁试剂网等市售途径购买获得的普通试剂。
[0059]
实施例1
[0060]
制备功能化聚碳酸酯,步骤为:
[0061]
(1)将94g(1mol)丙烯酸钠、618g(3mol)对苯乙烯磺酸钠、0.912g(0.004mol)过硫酸铵、412.32g(0.04mol)二烯丙基双酚a加入到11748g超纯水中,氮气氛围保护,75℃下预
聚合反应6h后,将温度降至室温,撤去氮气,在空气中暴露终止反应,得到预聚物。通过gpc测试,得到预聚物的重均分子量为3500。
[0062]
ftir测试结果显示1720cm-1
处出现了吸收峰,这归功于丙烯酸上的羧基;除此之外,1163cm-1
和1092cm-1
处出现了吸收峰,这要归于对苯乙烯磺酸上的磺酸基基团,在3350cm-1
处出现吸收峰,这是烯丙基双酚a的酚羟基的特征吸收峰,同时在1650cm-1
处观察不到吸收峰,表明双键消失,反生了聚合反应。
[0063]
(2)将在有氮气保护的反应器中加入64.13g预聚物(0.018mol),228.29g(1mol)双酚a,5g对叔丁基苯酚,188.34g氢氧化钠,3766.79超纯水混合,机械搅拌直至完全溶解得到澄清溶液,然后加入3766.79g二氯甲烷,机械搅拌形成稳定的水油混合体系。
[0064]
(3)向水油混合体系中加入0.046g催化剂三乙胺,同时按照0.94g/min的速率通入112.91g(1.14mol)光气进行聚合反应。整个反应过程中利用32wt%氢氧化钠水溶液维持体系ph为11-12,反应温度为30℃,反应时间120min。待反应结束后,经过20wt%naoh溶液碱洗、0.5mol/l盐酸酸洗、去离子水洗涤、脱挥(40℃玻璃釜中水煮制粉2h)、粉碎、干燥(120℃鼓风烘箱干燥4h)得到功能化聚碳酸酯产物。
[0065]
利用gpc测得产物的重均分子量为24300,多分散指数为1.58。
[0066]
核磁测试的结果显示预聚物的含量为25.1wt%。衰减全反射红外测试结果显示1163cm-1
和1092cm-1
处出现了吸收峰,这是磺酸基的典型吸收峰,在1720cm-1
处观察到羰基的吸收峰,同时在3350cm-1
处观察不到烯丙基双酚a的酚羟基的特征吸收峰。
[0067]
红外的测试结果表明制备得到本发明功能化聚碳酸酯。
[0068]
核磁结果显示其结构中m取值为7,n取值为3。
[0069]
实施例2
[0070]
制备功能化聚碳酸酯,步骤为:
[0071]
(1)将94g(1mol)丙烯酸钠、206g(1mol)对苯乙烯磺酸钠、0.238g(0.001mol)引发剂过硫酸钠、3.08g(0.01mol)二烯丙基双酚a加入到6000g超纯水中,氮气氛围保护,65℃下预聚合反应4h后,将温度降至室温,撤去氮气,在空气中暴露终止反应,得到预聚物。通过gpc测试,得到预聚物的重均分子量为1200。
[0072]
(2)将在有氮气保护的反应器中加入15.39g(0.013mol)预聚物,228.29g双酚a(1mol),5g封端剂苯酚,136.974g氢氧化钾,4565.8g超纯水混合,机械搅拌直至完全溶解得到澄清溶液,然后加入2282.9g惰性有机溶剂二氯乙烷,机械搅拌形成稳定的水油混合体系。
[0073]
(3)向水油混合体系中加入0.023g催化剂四丁基溴化铵,同时按照3.342g/min的速率通入100.27g(1.013mol)光气进行聚合反应。整个反应过程中利用32wt%氢氧化钠水溶液维持体系ph为11-12,反应温度为25℃,反应时间30min。待反应结束后,经过20wt%naoh溶液碱洗、0.5mol/l盐酸酸洗、去离子水洗涤、脱挥(40℃玻璃釜中水煮制粉2h)、粉碎、干燥(120℃鼓风烘箱干燥4h)得到功能化聚碳酸酯产物。
[0074]
利用gpc测得产物的重均分子量为23300,多分散指数为1.6。
[0075]
核磁测试结果显示含预聚物的含量为6.05%,
[0076]
红外的测试结果表明制备得到本发明功能化聚碳酸酯。
[0077]
核磁结果显示其结构中m取值为1,n取值为9。
[0078]
实施例3
[0079]
制备功能化聚碳酸酯,步骤为:
[0080]
(1)将94g(1mol)丙烯酸钠、1030g(5mol)对苯乙烯磺酸钠、16.2g(0.06mol)过硫酸钾、27.27g(0.09mol)二烯丙基双酚a加入到14050g超纯水中,氮气氛围保护,80℃下预聚合反应24h后,将温度降至室温,撤去氮气,在空气中暴露终止反应,得到预聚物。通过gpc测试,得到预聚物的重均分子量为10000。
[0081]
(2)将在有氮气保护的反应器中加入51.30g(0.005mol)预聚物,228.29g双酚a(1mol),5g对叔丁基苯酚,273.948g氢氧化钠,3424.35超纯水混合,机械搅拌直至完全溶解得到澄清溶液,然后加入3595.58g二氯甲烷,机械搅拌形成稳定的水油混合体系。
[0082]
(3)向水油混合体系中加入1.141g催化剂三乙胺,同时按照0.477g/min的速率通入114.43g(1.156mol)光气进行聚合反应。整个反应过程中利用32wt%氢氧化锂水溶液维持体系ph为11-12,反应温度为35℃,反应时间240min。待反应结束后,经过20wt%naoh溶液碱洗、0.5mol/l盐酸酸洗、去离子水洗涤、脱挥(40℃玻璃釜中水煮制粉2h)、粉碎、干燥(120℃鼓风烘箱干燥4h)得到功能化聚碳酸酯产物。
[0083]
利用gpc测得产物的重均分子量为24300,多分散指数为1.58。
[0084]
核磁测试的结果显示预聚物的含量为20.1wt%。
[0085]
红外的测试结果表明制备得到本发明功能化聚碳酸酯。
[0086]
核磁结果显示其结构中m取值为10,n取值为1。
[0087]
实施例4
[0088]
制备功能化聚碳酸酯,步骤为:
[0089]
(1)将94g(1mol)丙烯酸钠、412g(2mol)对苯乙烯磺酸钠、0.684g(0.003mol)过硫酸铵、7.98g(0.03mol)二烯丙基双酚s加入到7590g超纯水中,氮气氛围保护,70℃下预聚合反应7h后,将温度降至室温,撤去氮气,在空气中暴露终止反应,得到预聚物。通过gpc测试,得到预聚物的重均分子量为2800。
[0090]
(2)将在有氮气保护的反应器中加入119.27g(0.043mol)预聚物,268.35g(1mol)双酚z,5.56g对叔丁基苯酚,201.26g氢氧化钠,4025.25超纯水混合,机械搅拌直至完全溶解得到澄清溶液,然后加入4025.25g二氯甲烷,机械搅拌形成稳定的水油混合体系。
[0091]
(3)向水油混合体系中加入0.054g催化剂三乙胺,同时按照0.963g/min的速率通入115.6g(1.167mol)光气进行聚合反应。整个反应过程中利用32wt%氢氧化钠水溶液维持体系ph为11-12,反应温度为30℃,反应时间120min。待反应结束后,经过20wt%naoh溶液碱洗、0.5mol/l盐酸酸洗、去离子水洗涤、脱挥(40℃玻璃釜中水煮制粉2h)、粉碎、干燥(120℃鼓风烘箱干燥4h)得到功能化聚碳酸酯产物。
[0092]
利用gpc测得产物的重均分子量为24965,多分散指数为1.62。
[0093]
核磁测试的结果显示预聚物的含量为40wt%。
[0094]
红外的测试结果表明制备得到本发明功能化聚碳酸酯。
[0095]
核磁结果显示其结构中m取值为5,n取值为5。
[0096]
实施例5
[0097]
参照实施例1方法制备功能化聚碳酸酯:不同之处仅在于:封端剂对叔丁基苯酚的加入量为4.4g,其余操作与实施例1相同。
[0098]
利用gpc测得产物的重均分子量为28560,多分散指数为1.63。核磁测试的结果显示产物a的含量为25.05wt%,其结构中m取值为4,n取值为6。
[0099]
实施例6
[0100]
参照实施例2方法制备功能化聚碳酸酯:不同之处仅在于封端剂对叔丁基苯酚的加入量为4.4g。其余操作与实施例2相同。利用gpc测得产物的重均分子量为29200,多分散指数为2.05。核磁测试的结果显示产物a的含量为6.03wt%,其结构中m取值为8,n取值为2。
[0101]
实施例7
[0102]
参照实施例3方法制备功能化聚碳酸酯:不同之处仅在于封端剂对叔丁基苯酚的加入量为3.4g。其余操作与实施例3相同。利用gpc测得产物的重均分子量为36200,多分散指数为1.61。核磁测试的结果显示产物a的含量为20.1wt%,其结构中m取值为1,n取值为10。
[0103]
对比例1
[0104]
制备聚碳酸酯,步骤为:
[0105]
将在有氮气保护的反应器中加入228.29g双酚a,5g对叔丁基苯酚,188.34g氢氧化钠,3766.785g超纯水混合,机械搅拌直至完全溶解,然后加入3766.785g二氯甲烷,机械搅拌形成稳定的水油混合体系。之后加入0.046g催化剂三乙胺,同时将110.88g光气按照0.924g/min的速率通入到上述体系中。整个反应过程中加入32wt%氢氧化钠水溶液维持体系ph为11-12,反应温度为30℃。待反应结束后,经过后处理(处理方法同实施例1)得到产物。
[0106]
利用gpc测得产物的重均分子量为24520,多分散指数为1.71。衰减全反射红外光谱显示在1611cm-1
、1509cm-1
、1446cm-1
处有明显的吸收峰,这是苯环的特征峰,没有观察到羧基及磺酸基的典型吸收峰。
[0107]
对比例2
[0108]
参照实施例1方法制备聚碳酸酯,不同之处仅在于:步骤1)中不加入丙烯酸钠单体,其他操作及参数均不变,制得聚碳酸酯。
[0109]
对比例3
[0110]
参照实施例1方法制备聚碳酸酯,不同之处仅在于:步骤1)中丙烯酸钠单体替换为反式-2己烯酸,其他操作及参数均不变,制得聚碳酸酯。
[0111]
对比例4
[0112]
参照实施例1方法制备聚碳酸酯,不同之处仅在于:步骤1)中不加入对苯乙烯磺酸钠单体,其他操作及参数均不变,制得聚碳酸酯。
[0113]
对比例5
[0114]
参照实施例1方法制备聚碳酸酯,不同之处仅在于:步骤1)中对苯乙烯磺酸钠单体替换为苯基乙烯基砜单体,其他操作及参数均不变,制得聚碳酸酯。
[0115]
对比例6
[0116]
参照实施例1方法制备聚碳酸酯,不同之处在于:步骤3)中不加入催化剂。
[0117]
以上各实施例和对比例的制备的聚碳酸酯性能测试结果如下表1所示:
[0118]
表1
[0119][0120][0121]
通过以上数据对比可知,相比常规聚碳酸酯,本发明的聚碳酸酯共聚物的力学性能及流动性能基本得以保持,血液相容性提高,尤其具有自抗凝性,表现为atpp、tt时间明显延长、溶血率显著降低,血小板激活激活因子的浓度明显降低,能够有效抑制血小板的激活,有效地提升了聚碳酸酯的性能,拓宽了材料的应用领域。
[0122]
本领域技术人员可以理解,在本说明书的教导之下,可对本发明做出一些修改或调整。这些修改或调整也应当在本发明权利要求所限定的范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。