1.本发明涉及有机材料领域,特别涉及一种含氟二胺单体、聚酰亚胺、聚酰亚胺薄膜以及制备方法。
背景技术:
2.传统的聚酰亚胺材料(pi)一般采用芳香族的二酐单体和二胺单体通过两步法或一步法缩合聚合制得,由于分子内和分子间电荷转移络合物(ctc)的形成,全芳香族pi薄膜一般呈现棕黄色,在紫外可见光区(波长400~800nm)透过率低下,400nm处几乎被100%吸收,无法满足光电领域如光通讯领域中的光波导材料,光电材料以及液晶显示领域的取向膜材料等应用需求。
3.为了改善pi薄膜的透光性能,研究人员采取了多种策略,如1)引入氟原子,利用氟原子电负性大,c-f键极化率小的特点帮助降低ctc的形成从而改善透光性;2)采用含大侧基或非共面扭曲结构的二酐二胺单体,减弱聚酰亚胺分子链的有序排列,减弱二酐二胺共轭性从而阻碍ctc的形成改善透光性,如采用含苯基,叔丁基,萘,联萘等结构的二酐或二胺单体;3)采用脂肪/脂环族单体,由于脂环结构不存在π电子,不利于ctc的形成从而改善透光性能。虽然理论上,上述各种改进方式会改善pi薄膜的透光性,但是有机结构的构效关系密切,实际合成及应用中,结构的改变对材料性能的影响比较复杂,比如并非任意结构的二胺中引入f原子或f原子以任意形式引入二胺,都能有效改善材料的透光性,再如采用脂环族单体来试图改善材料的透光性,但是会导致聚酰亚胺的耐热性能下降。因此,在实际应用中需要综合考量薄膜的透光性和耐热性,研制具有高耐热性高透光性的pi膜也成为许多科研工作者的目标。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种含氟二胺单体、聚酰亚胺、聚酰亚胺薄膜以及制备方法。采用本发明的二胺单体制备的聚酰亚胺不仅能够提升材料的透光性,还能改善材料的耐热性能。
5.本发明提供了一种含氟二胺单体,具有式(ⅰ)所示结构:
[0006][0007]
其中:
[0008]rf1
、r
f2
独立的选自:-h、-f或-cf3;且r
f1
和r
f2
中至少有1个不为-h;
[0009]
基团为含降冰片二烯基的基团。
[0010]
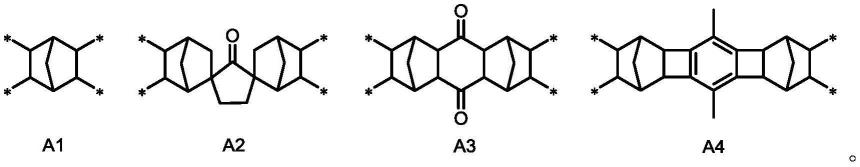
优选的,基团选自式a1~式a4所示结构:
[0011][0012]
优选的,选自以下结构:
[0013]
[0014][0015]
本发明还提供了一种上述技术方案中所述的含氟二胺单体的制备方法,包括以下步骤:
[0016]
在保护性气氛中,在过渡金属基催化剂、有机磷配体和无机碱性物质存在的条件下,卤代苯胺和降冰片二烯类化合物反应,形成式(ⅰ)所示的含氟二胺单体;
[0017]
其中:
[0018]
所述降冰片二烯类化合物为降冰片二烯和降冰片二烯衍生物中的一种或几种;
[0019]
所述卤代苯胺为取代的卤代苯胺,取代基选自:-f和/或-cf3。
[0020]
优选的,所述降冰片二烯类化合物选自式1-1~式1-4所示化合物中的一种或几种:
[0021][0022]
所述卤代苯胺选自式2-1~式2-13所示化合物中的一种或几种:
[0023][0024]
其中,x选自:-br或-cl。
[0025]
优选的,所述过渡金属基催化剂选自三(二亚苄基丙酮)二钯、双二亚苄基丙酮钯、醋酸钯、三氟乙酸钯、氯化钯、双乙酰丙酮钯、六氟乙酰丙酮钯、(1,5-环辛二烯)二氯化钯、氯化烯丙基钯二聚物、二(三叔丁基膦)钯、四(三苯基膦)钯、双[三(2-甲苯基)膦]合钯、双乙腈氯化钯、二碘化钯、乙酰丙酮钯、[1,1'-双(二苯基膦)二茂铁]二氯化钯、双三苯基磷二氯化钯中的一种或几种;
[0026]
所述有机磷配体选自三苯基膦、三(对甲基苯基)膦、三(邻甲基苯基)磷、三苄基膦、三环己基膦、三叔丁基膦、4,5-双二苯基膦-9,9-二甲基氧杂蒽、2-二环己基磷-2',6'-二异丙氧基-1,1'-联苯、1,2-双(二苯基膦基)苯、1,1'-双(二苯基膦)二茂铁、1,2-双(二苯基膦)乙烷、1,1'-联萘-2,2'-双二苯膦、2-二环己膦基-2'-(n,n-二甲胺)-联苯中的一种或几种;
[0027]
所述无机碱性物质选自磷酸钾、磷酸钠、磷酸氢钾、磷酸氢钠、碳酸铯、碳酸钾、碳酸钠、碳酸锂、碳酸氢钠、碳酸氢钾、氟化铯、氟化钾、叔丁醇锂、叔丁醇钠、叔丁醇钾、苯酚钠、苯酚钾、氢氧化钠、氢氧化钾中的一种或几种。
[0028]
本发明还提供了一种聚酰亚胺,形成所述聚酰亚胺采用的二胺单体原料包括上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体。
[0029]
本发明还提供了一种上述技术方案中所述的聚酰亚胺的制备方法,包括以下步骤:
[0030]
在保护性气氛中,二胺单体和二酐单体反应,形成聚酰亚胺共聚物。
[0031]
所述二胺单体为上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体。
[0032]
优选的,所述二酐单体选自式
ⅲ‑
1~式
ⅲ‑
6所示化合物中的一种或几种:
[0033][0034]
本发明还提供了一种聚酰亚胺薄膜,所述薄膜中的聚酰亚胺为上述技术方案中所述的聚酰亚胺或为上述技术方案中所述的制备方法制得的聚酰亚胺。
[0035]
本发明提供的含氟二胺单体,具有式(ⅰ)所示结构,其具有含氟基团(-f和/或-cf3)以及双苯并环丁烷基团,上述特定的结构的二胺单体能够使聚酰亚胺薄膜具有较高的透光性,同时使材料具有更好的耐热性和尺寸稳定性。
[0036]
实验结果表明,本发明提供的含氟二胺单体,能够使聚酰亚胺在400nm处的透光率≥85%,截止波长λ
cut-off
≤290nm;玻璃化转变温度≥390℃;热膨胀系数cte在25ppmk-1
以下。
附图说明
[0037]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
[0038]
图1为实施例1中制备的二胺单体产物中
ⅰ‑
syn-1二胺单体的核磁氢谱图;
[0039]
图2为实施例2中制备的二胺单体产物中
ⅰ‑
anti-5二胺单体的核磁氢谱图;
[0040]
图3为实施例3中制备的二胺单体
ⅰ‑
syn-6和i-anti-6的核磁氢谱图;
[0041]
图4为实施例4中制备的二胺单体
ⅰ‑
syn-11和
ⅰ‑
anti-11的核磁氢谱图;
[0042]
图5为实施例5中制备的二胺单体
ⅰ‑
syn-21和
ⅰ‑
anti-21的核磁氢谱图;
[0043]
图6为实施例6中制备的二胺单体
ⅰ‑
syn-25和
ⅰ‑
anti-25的核磁氢谱图;
[0044]
图7为实施例6中制备的聚酰亚胺共聚物的核磁氢谱图;
[0045]
图8为对实施例1~6所得聚酰亚胺薄膜的动态热机械分析(dma)曲线图;
[0046]
图9为实施例1~6所得聚酰亚胺薄膜的失重(tga)曲线图;
[0047]
图10为实施例1~6所得聚酰亚胺薄膜的可见光区光学透过性能示意图。
具体实施方式
[0048]
本发明提供了一种含氟二胺单体,具有式(ⅰ)所示结构:
[0049][0050]
其中:
[0051]rf1
、r
f2
独立的选自:-h、-f或-cf3;且r
f1
和r
f2
中至少有1个不为-h;r
f1
和r
f2
在母环上的取代位置没有特殊限制;
[0052]
基团为含降冰片二烯基的基团。
[0053]
优选的,基团选自式a1~式a4所示结构:
[0054][0055]
其中,*代表连接位置。
[0056]
本发明中,所述式(ⅰ)所示含氟二胺单体优选具有顺式结构(即syn式结构)和/或反式结构(即anti式结构);更优选具有顺式结构和反式结构。所述顺式结构和反式结构的摩尔比优选为(1~5)∶1。
[0057]
其中,基团为式a1时,所述式(ⅰ)所示含氟二胺单体优选为以下顺式结构
ⅰ‑
syn-1~
ⅰ‑
syn-10和反式结构
ⅰ‑
anti-1~
ⅰ‑
anti-10中的一种或几种:
[0058][0059]
具体的:基团为式a1时,卤代苯胺为式2-1时,所述
ⅰ‑
syn-1和
ⅰ‑
anti-1的摩尔比优选为5∶1。基团为式a1时,卤代苯胺为式2-3时,所述
ⅰ‑
syn-2和
ⅰ‑
anti-2的摩尔比
优选为2∶1。基团为式a1时,卤代苯胺为式2-2时,所述
ⅰ‑
syn-3和
ⅰ‑
anti-3的摩尔比优选为1∶1。基团为式a1时,卤代苯胺为式2-4时,所述
ⅰ‑
syn-4和
ⅰ‑
anti-4的摩尔比优选为1∶1。基团为式a1时,卤代苯胺为式2-5时,所述
ⅰ‑
syn-5和
ⅰ‑
anti-5的摩尔比优选为1.68∶1。基团为式a1时,卤代苯胺为式2-6时,所述
ⅰ‑
syn-6和
ⅰ‑
anti-6的摩尔比优选为1∶1。基团为式a1时,卤代苯胺为式2-7时,所述
ⅰ‑
syn-7和
ⅰ‑
anti-7的摩尔比优选为1∶1。基团为式a1时,卤代苯胺为式2-10时,所述
ⅰ‑
syn-8和
ⅰ‑
anti-8的摩尔比优选为1∶1。基团为式a1时,卤代苯胺为式2-11时,所述
ⅰ‑
syn-9和
ⅰ‑
anti-9的摩尔比优选为1∶1。基团为式a1时,卤代苯胺为式2-13时,所述
ⅰ‑
syn-10和
ⅰ‑
anti-10的摩尔比优选为1∶1。
[0060]
其中,基团为式a2时,所述式(ⅰ)所示含氟二胺单体优选为以下顺式结构
ⅰ‑
syn-11~
ⅰ‑
syn-16和反式结构
ⅰ‑
anti-11~
ⅰ‑
anti-16中的一种或几种:
[0061][0062]
具体的:基团为式a2时,所述顺式结构和反式结构的摩尔比优选为1∶1。
[0063]
其中,基团为式a3时,所述式(ⅰ)所示含氟二胺单体优选为以下顺式结构
ⅰ‑
syn-17~
ⅰ‑
syn-22和反式结构
ⅰ‑
anti-17~
ⅰ‑
anti-22中的一种或几种:
[0064][0065]
具体的:基团为式a3时,所述顺式结构和反式结构的摩尔比优选为1∶1。
[0066]
其中,基团为式a4时,所述式(ⅰ)所示含氟二胺单体优选为以下顺式结构
ⅰ‑
syn-23~
ⅰ‑
syn-28和反式结构
ⅰ‑
anti-23~
ⅰ‑
anti-28中的一种或几种:
[0067][0068]
具体的:基团为式a4时,所述顺式结构和反式结构的摩尔比优选为1∶1。
[0069]
本发明提供的含氟二胺单体,具有式(ⅰ)所示结构,其具有含氟基团(-f、-cf3)以及双苯并环丁烷基团,上述特定的结构能够使聚酰亚胺薄膜具有较高的透光性,同时使材料具有更好的耐热性。
[0070]
实验结果表明,本发明提供的含氟二胺单体,能够使聚酰亚胺在400nm处的透光率≥85%,截止波长λ
cut-off
≤290nm;玻璃化转变温度≥390℃;热膨胀系数cte在25ppmk-1
以下。
[0071]
本发明还提供了一种上述技术方案中所述的含氟二胺单体的制备方法,包括以下步骤:在保护性气氛中,在过渡金属基催化剂、有机磷配体和无机碱性物质存在的条件下,卤代苯胺和降冰片二烯类化合物反应,形成式(ⅰ)所示的含氟二胺单体;
[0072]
其中:
[0073]
所述降冰片二烯类化合物为降冰片二烯和降冰片二烯衍生物中的一种或几种;
[0074]
所述卤代苯胺为取代的卤代苯胺,取代基选自:-f和/或-cf3。
[0075]
本发明中,所述保护性气氛的气体种类没有特殊限制,为本领域技术人员熟知的常规惰性气体即可,如氮气或氩气等。
[0076]
本发明中,所述过渡金属基催化剂优选为三(二亚苄基丙酮)二钯、双二亚苄基丙酮钯、醋酸钯、三氟乙酸钯、氯化钯、双乙酰丙酮钯、六氟乙酰丙酮钯、(1,5-环辛二烯)二氯化钯、氯化烯丙基钯二聚物、二(三叔丁基膦)钯、四(三苯基膦)钯、双[三(2-甲苯基)膦]合钯、双乙腈氯化钯、二碘化钯、乙酰丙酮钯、[1,1'-双(二苯基膦)二茂铁]二氯化钯、双三苯基磷二氯化钯中的一种或几种。
[0077]
本发明中,所述有机磷配体优选为三苯基膦、三(对甲基苯基)膦、三(邻甲基苯基)磷、三苄基膦、三环己基膦、三叔丁基膦、4,5-双二苯基膦-9,9-二甲基氧杂蒽、2-二环己基磷-2',6'-二异丙氧基-1,1'-联苯、1,2-双(二苯基膦基)苯、1,1'-双(二苯基膦)二茂铁、1,2-双(二苯基膦)乙烷、1,1'-联萘-2,2'-双二苯膦、2-二环己膦基-2'-(n,n-二甲胺)-联苯中的一种或几种。
[0078]
本发明中,所述无机碱性物质优选为磷酸钾、磷酸钠、磷酸氢钾、磷酸氢钠、碳酸铯、碳酸钾、碳酸钠、碳酸锂、碳酸氢钠、碳酸氢钾、氟化铯、氟化钾、叔丁醇锂、叔丁醇钠、叔丁醇钾、苯酚钠、苯酚钾、氢氧化钠、氢氧化钾中的一种或几种。
[0079]
本发明中,所述卤代苯胺为取代的卤代苯胺,取代基选自:-f和/或-cf3。优选的,所述卤代苯胺选自式2-1~式2-13所示化合物中的一种或几种:
[0080][0081]
其中,x选自:-br或-cl。在本发明的一些实施例中,卤代苯胺为4-溴-3-三氟甲基苯胺、4-氯-2-三氟甲基苯胺、4-氯-2-氟-5-三氟甲基苯胺或4-溴2,5-二氟苯胺。
[0082]
本发明中,所述降冰片二烯类化合物为降冰片二烯和降冰片二烯衍生物中的一种或几种。优选为式1-1~式1-4所示化合物中的一种或几种:
[0083][0084]
其中,式1-1(即nbd,降冰片二烯)的来源没有特殊限制,为一般市售品即可。式1-2(即nscsn,2-螺环-α-环戊酮-α
’‑
螺环-2
”‑5”‑
降冰片烯)、式1-3(即dmoha,类蒽醌结构的双降冰片烯)和式1-4(即npnbd,含苯并环丁烷结构的双降冰片烯)按照已公开专利文献或非专利文献报道的合成方法合成。所制备的式1-2、式1-2和式1-2为多种空间异构体(endo/exo)的混合物,本发明采用的便是这种未加分离的异构体混合物。
[0085]
本发明中,所述卤代苯胺与降冰片二烯类化合物的摩尔比优选为2∶1。本发明中,所述过渡金属基催化剂与降冰片二烯类化合物的摩尔比优选为(2~20)∶1。所述有机磷配体与降冰片二烯类化合物的摩尔比优选为(4~40)∶1。所述碱性物质与降冰片二烯类化合物的摩尔比优选为(2~4)∶1。
[0086]
本发明中,上述反应在有机溶剂介质中进行。所述有机溶剂优选为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、1,3-二甲基-3,4,5,6-四氢-2-嘧啶酮、n-甲基吡咯烷酮、二甲基亚砜、1,4-二氧六环、2-甲基四氢呋喃、苯甲醚、对二氯苯、甲苯和三氟甲苯中的一种或几种。本发明中,所述有机溶剂与降冰片二烯类化合物的用量比优选为(2~200)ml∶(2~50)mmol。
[0087]
本发明中,所述反应的温度优选为100~200℃;所述反应的时间优选为24~72h。本发明中,经上述反应后,形成含氟二胺单体。
[0088]
本发明中,所得二胺单体为反式结构(
ⅰ‑
anti)和/或顺式结构式(
ⅰ‑
syn),更优选为反式结构(
ⅰ‑
anti)和顺式结构式(
ⅰ‑
syn)两种位置异构体的混合物。本发明中,所述包含反式结构(
ⅰ‑
anti)和顺式结构式(
ⅰ‑
syn)的二胺单体中,反式结构(
ⅰ‑
anti)与顺式结构式(
ⅰ‑
syn)的摩尔比为(1~5)∶1。本发明在得到含顺反异构体的二胺单体后,可通过柱层析分
离,获得单一结构的二胺单体,将单一结构的二胺单体用于聚酰亚胺的聚合反应;也可以不做分离,直接采用含有顺反异构体混合物的二胺单体,进行聚酰亚胺的聚合反应。
[0089]
本发明提供的上述制备方法原料易得、反应步骤简单(一步反应制得),且产品收率较高(大于80%),有利于规模化生产应用。
[0090]
本发明还提供了一种聚酰亚胺,形成所述聚酰亚胺采用的二胺单体原料包括上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体。以其作为反应原料制得的聚酰亚胺共聚物,不仅能够提升材料的光学性能,还能提高材料的耐热性。
[0091]
本发明还提供了一种上述技术方案中所述的聚酰亚胺的制备方法,包括以下步骤:
[0092]
在保护性气氛中,二胺单体和二酐单体反应,形成聚酰亚胺共聚物;
[0093]
所述二胺单体为上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体。
[0094]
具体的,所述制备方法可以为一步法或两步法。
[0095]
关于一步法,包括以下步骤:
[0096]
在保护性气氛下,二胺单体和二酐单体在有机溶剂中,在一定温度下搅拌至溶解后,升高温度继续反应得到聚酰亚胺共聚物。
[0097]
本发明中,提供所述保护性气氛的气体种类没有特殊限制,为本领域技术人员熟知的常规保护性气体即可,如氮气或氩气等。
[0098]
本发明中,所述二胺单体为上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体,在此不再赘述。
[0099]
本发明中,所述二酐单体优选为式
ⅲ‑
1~式
ⅲ‑
6所示化合物中的一种或几种:
[0100][0101]
本发明中,所述二胺单体与二酐单体的摩尔比优选为(0.95~1.05)∶1。
[0102]
本发明中,所述反应可在催化剂的作用下进行。所述催化剂包括碱性催化剂和/或酸性催化剂。其中,所述碱性催化剂优选为异喹啉和/或三乙胺。所述酸性催化剂优选为苯甲酸和/或对羟基苯甲酸。本发明中,所述催化剂与二胺单体的摩尔比优选为(0.1~0.5):1。
[0103]
本发明中,所述有机溶剂优选为极性非质子溶剂,更优选为间甲酚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等中的一种或多种。
[0104]
本发明中,所述二胺单体在所述有机溶剂中的初始固含量优选为10wt%~30wt%。所述初始固含量是指仅将二胺单体加入溶剂中、还未加入其它单体原料时的固含
量。
[0105]
本发明中,所述溶解温度优选为40~80℃;所述溶解时间优选为1~12h。所述反应的温度优选为150~200℃;所述反应时间优选为6~48h。本发明中,经上述反应后,得到粘稠的聚酰亚胺溶液。本发明中,所述聚酰亚胺溶液的固含量优选为15wt%~30wt%。
[0106]
关于两步法,包括以下步骤:
[0107]
a)在保护性气氛中,二胺单体和二酐单体在有机溶剂中反应,得到聚酰胺酸溶液;
[0108]
b)对所述聚酰胺酸溶液进行酰亚胺化处理,得到聚酰亚胺共聚物;
[0109]
所述二胺单体为上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体。
[0110]
关于步骤a):在保护性气氛中,二胺单体和二酐单体在有机溶剂中反应,得到聚酰胺酸溶液。
[0111]
本发明中,提供所述保护性气氛的气体种类没有特殊限制,为本领域技术人员熟知的常规保护性气体即可,如氮气或氩气等。
[0112]
本发明中,所述二胺单体为上述技术方案中所述的含氟二胺单体或由上述技术方案中所述的制备方法制得的含氟二胺单体,在此不再赘述。
[0113]
本发明中,所述二酐单体优选为式
ⅲ‑
1~式
ⅲ‑
6所示化合物中的一种或几种:
[0114][0115]
本发明中,所述二胺单体与二酐单体的摩尔比优选为(0.95~1.05)∶1。
[0116]
本发明中,所述反应可在催化剂的作用下进行。所述催化剂包括碱性催化剂和/或酸性催化剂。其中,所述碱性催化剂优选为异喹啉和/或三乙胺。所述酸性催化剂优选为苯甲酸和/或对羟基苯甲酸。本发明中,所述催化剂与二胺单体的摩尔比优选为(0.1~0.5):1。
[0117]
本发明中,所述有机溶剂优选为极性非质子溶剂,更优选为间甲酚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等中的一种或多种。本发明中,所述二胺单体在所述有机溶剂中的初始固含量优选为10wt%~30wt%。所述初始固含量是指仅将二胺单体加入溶剂中、还未加入其它单体原料时的固含量。
[0118]
本发明中,所述反应的温度优选为0~50℃;所述反应的时间优选为6~48h。本发明中,经上述反应后,得到粘稠的聚酰胺酸溶液。本发明中,所述聚酰胺酸溶液的固含量优选为10wt%~30wt%。
[0119]
关于步骤b):对所述聚酰胺酸溶液进行酰亚胺化处理,得到聚酰亚胺共聚物。
[0120]
本发明中,所述酰亚胺化处理包括化学亚胺化处理和热亚胺化处理。
[0121]
所述热亚胺化:是指在高温下进行热处理,使聚酰胺酸发生亚胺化转变。本发明
中,所述热亚胺化的温度优选为150~200℃;所述热亚胺化的时间优选为6~24h。
[0122]
所述化学亚胺化:是指在化学添加剂(如催化剂、脱水剂等)的作用下,使聚酰胺酸发生亚胺化转变。本发明中,所述化学添加剂为催化剂和脱水剂。其中,所述催化剂优选为p-吡咯啉、二甲基吡啶、三甲基吡啶、喹啉、异喹啉和三乙胺中的一种或多种。所述脱水剂优选为三氟乙酸酐、乙酸酐和丙酸酐中的一种或多种。本发明中,所述化学添加剂的种类及用量没有特殊限制,为本领域常规种类及用量即可。
[0123]
本发明经化学酰亚胺化处理后,体系中聚酰胺酸转变为聚酰亚胺共聚物。本发明中,经上述化学酰亚胺化处理后,优选还进行纯化后处理。所述纯化的方式优选为:将体系中形成的沉淀物在索式提取器中,采用醇类溶剂,索提回流12~72h;或将沉淀物重新溶解,倒入醇类溶剂中再次析出,过滤,并重复2~3次。其中,所述醇类溶剂优选为甲醇和/或乙醇。经上述后处理后,得到聚酰亚胺共聚物。
[0124]
以两步法为例,二胺单体与二酐单体反应形成聚酰亚胺的合成路线如下:
[0125][0126]
本发明制得的聚酰亚胺具有式(ⅱ)所示结构:
[0127][0128]
其中:
[0129]
ra为酸酐基团,优选的,ra选自式3-1~式3-6所示结构:
[0130][0131]
rb对应式(ⅰ)二胺单体中的连接于两氨基之间的基团,具体参见前文,在此不再赘述。
[0132]
本发明还提供了一种聚酰亚胺薄膜,所述薄膜中的聚酰亚胺为上述技术方案中所述的聚酰亚胺或为上述技术方案中所述的制备方法制得的聚酰亚胺。即本发明经上述技术方案中所述的聚酰亚胺共聚物作为原料,通过制膜手段成膜,得到聚酰亚胺薄膜。
[0133]
本发明中,所述制膜的方法没有特殊限制,为本领域常规制膜方式即可。本发明中,所述制膜的方法优选具体为:
[0134]
s1、将所述聚酰亚胺溶解于溶剂中,得到聚酰亚胺溶液;
[0135]
s2、将所述聚酰亚胺溶液涂覆于支撑体上,干燥,形成聚酰亚胺湿膜;
[0136]
s3、将所述聚酰亚胺湿膜从支撑体上剥离、干燥和拉伸处理,得到聚酰亚胺薄膜。
[0137]
其中,所述溶剂优选为有机溶剂,更优选包括四氢呋喃、二氯甲烷、三氯甲烷、间甲酚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮和γ-丁内酯中的一种或多种。所述聚酰亚胺溶液的固含量优选为5wt%~15wt%。所述步骤s2中,优选采用流延法使聚酰亚胺溶液均匀涂覆于支撑体上;所述支撑体为洁净的支撑体。所述步骤s2中,所述干燥的温度优选为100~300℃。
[0138]
本发明中,所述聚酰亚胺薄膜的厚度优选为20~50μm。
[0139]
本发明所提供的含氟高透明聚酰亚胺薄膜用于柔性显示、薄膜太阳能电池或光电子工程领域中。
[0140]
与现有技术相比,本发明具有以下有益效果:
[0141]
(1)本发明式(i)所示含氟二胺单体能够使聚酰亚胺薄膜具有优异的光学性能,在400nm处光学透过率≥85%,截止波长λ
cut-off
≤290nm;更好的耐热性能,其玻璃化转变温度≥390℃,可高可达481℃;良好的尺寸稳定性,热膨胀系数cte在25ppmk-1
以下。
[0142]
(2)本发明首次将含氟基团、双苯并环丁烷基团的二胺单体应用于制备无色聚酰亚胺薄膜,本发明中含氟二胺单体的制备方法原料易得,反应步骤简单(一步反应制得),收率较高(大于80%)。本发明制备主链含脂环结构的含氟高透明聚酰亚胺及其薄膜具有优异的光学性能及良好的热性能等优点,在柔性显示,薄膜太阳能电池,光电子工程等领域有很好的应用前景。
[0143]
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
[0144]
以下实施例中所采用的实验材料,如无特殊说明,均可由常规的生化试剂公司购买得到。以下实施例中,室温是指25℃。
[0145]
实施例1
[0146]
1、制备二胺单体
[0147]
向反应瓶中加入醋酸钯(44.90mg,0.20mmol)、三苯基膦(104.92mg,0.40mmol)、碳酸铯(13.03g,40mmol)和无水1,4-二氧六环(40ml),室温搅拌10min后,依次加入式2-1所示4-溴-3-三氟甲基苯胺(9.56g,40mmol)、式1-1降冰片二烯(nbd)(1.84g,20mmol)继续搅拌若干分钟后,转移至150℃反应器中反应24h。待冷却至室温后,过滤除去不溶的无机盐,用乙酸乙酯淋洗,滤液浓缩后得粗产物。得到的粗产物通过快速柱层析提纯,得白色
ⅰ‑
1二胺固体7.30g,产率89%。i-1为syn式与anti式两种位置异构体的混合物(即
ⅰ‑
syn-1和
ⅰ‑
anti-1)。
[0148]
上述二胺单体的合成路线如下:
[0149][0150]
所得产物通过柱层析分离,分别得到
ⅰ‑
syn-1单体和
ⅰ‑
anti-1单体,
ⅰ‑
syn-1∶
ⅰ‑
anti-1的摩尔比为5∶1。其中,
ⅰ‑
syn-1二胺单体的核磁氢谱参见图1,图1为实施例1中制备
的二胺单体产物中
ⅰ‑
syn-1二胺单体的核磁氢谱图。
[0151]
2、制备聚酰亚胺薄膜
[0152]
在氩气氛围下,将未进行异构体分离的二胺单体
ⅰ‑
1(4.10g,10.00mmol)溶于无水n,n-二甲基乙酰胺中,初始固含量为30wt%,室温搅拌至二胺单体完全溶解。然后在相同温度下加入式
ⅲ‑
6所示二酐单体(即6fcda)(4.58g,10.00mmol),充分搅拌后,制得粘稠的聚酰胺酸溶液。在上述制备的聚酰胺酸溶液中加入吡啶(1.70ml,20.57mmol)和醋酸酐(2.00ml,21.02mmol)进行化学亚胺化。充分搅拌反应后,用甲醇沉淀出聚合物,将沉淀物在索式提取器中用甲醇作溶剂,索提回流48h后干燥得到聚酰亚胺共聚物。
[0153]
将所得聚酰亚胺共聚物溶解在n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0154]
实施例2
[0155]
1、制备二胺单体
[0156]
向反应瓶中加入氯化钯(35.47mg,0.20mmol)、1,1'-双(二苯基膦)二茂铁(221.75mg,0.40mmol)、氟化铯(6.08g,40mmol)和无水n,n-二甲基甲酰胺(40ml),室温搅拌10min后,依次加入式2-5所示4-氯-2-氟-5-三氟甲基苯胺(8.52g,40mmol)、式1-1降冰片二烯(nbd)(1.84g,20mmol)继续搅拌若干分钟后,转移至150℃反应器中反应24h。待冷却至室温后,过滤除去不溶的无机盐,用乙酸乙酯淋洗,滤液浓缩后得粗产物。得到的粗产物通过快速柱层析提纯,得白色
ⅰ‑
5二胺固体7.58g,产率85%。
ⅰ‑
5为syn式与anti式两种位置异构体的混合物(即
ⅰ‑
syn-5和
ⅰ‑
anti-5)。
[0157]
上述二胺单体的合成路线如下:
[0158][0159]
所得产物通过柱层析分离,分别得到i-syn-5单体和
ⅰ‑
anti-5单体,i-syn-5∶i-anti-5的摩尔比为1.68∶1。其中,i-anti-5二胺单体的核磁氢谱参见图1,图1为实施例2中制备的二胺单体产物中i-anti-5二胺单体的核磁氢谱图。
[0160]
2、制备聚酰亚胺薄膜
[0161]
在氩气氛围下,将未进行异构体分离的二胺单体i-5(4.46g,10.00mmol)、式
ⅲ‑
2所示二酐单体(即h-pmda)(2.24g,10.00mmol)和苯甲酸(0.24g,2.00mmol)溶于无水间甲酚中,固含量为30wt%,80℃搅拌至固体完全溶解。然后升温至180℃,充分搅拌反应后制得粘稠的聚酰亚胺溶液。降温后用甲醇沉淀出聚合物,将沉淀物在索式提取器中用甲醇作溶剂,索提回流48h后干燥得到聚酰亚胺共聚物。
[0162]
将所得聚酰亚胺共聚物溶解在n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0163]
实施例3
[0164]
1、制备二胺单体
[0165]
向反应瓶中加入三(二亚苄基丙酮)二钯(183.14mg,0.20mmol)、三环己基膦(112.17mg,0.40mmol)、苯酚钠(4.64g,40mmol)和无水n,n-二甲基乙酰胺(40ml),室温搅拌10min后,依次加入式2-6所示4-溴2,5-二氟苯胺(8.28g,40mmol)、式1-1降冰片二烯(nbd)(1.84g,20mmol)继续搅拌若干分钟后,转移至150℃反应器中反应48h。待冷却至室温后,过滤除去不溶的无机盐,用乙酸乙酯淋洗,滤液浓缩后得粗产物。得到的粗产物通过快速柱层析提纯,得白色
ⅰ‑
6二胺固体5.95g,产率86%。
ⅰ‑
6为syn式与anti式两种位置异构体的混合物(即
ⅰ‑
syn-6和
ⅰ‑
anti-6),
ⅰ‑
syn-6∶
ⅰ‑
anti-6的摩尔比为1∶1。
[0166]
所得产物的核磁氢谱参见图3,图3为实施例3中制备的二胺单体
ⅰ‑
syn-6和
ⅰ‑
anti-6的核磁氢谱图。
[0167]
上述二胺单体的合成路线如下:
[0168][0169]
2、制备聚酰亚胺薄膜
[0170]
在氩气氛围下,将所得二胺单体i-6(3.46g,10.00mmol)、式
ⅲ‑
3所示二酐单体(即cpoda)(3.84g,10.00mmol)和苯甲酸(0.24g,2.00mmol),溶于无水间甲酚中,固含量为30wt%,80℃搅拌至固体完全溶解。然后升温至180℃,加入异喹啉(0.26g,2.00mmol),充分搅拌后,制得粘稠的聚酰亚胺溶液。降温后用甲醇沉淀出聚合物,将沉淀物在索式提取器中用甲醇作溶剂,索提回流48h后干燥得到聚酰亚胺共聚物。
[0171]
将所得聚酰亚胺共聚物溶解在n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0172]
实施例4
[0173]
1、制备二胺单体
[0174]
向反应瓶中加入醋酸钯(89.80mg,0.40mmol)、三苯基膦(209.84mg,0.80mmol)、碳酸铯(13.03g,40mmol)与无水1,4-二氧六环,室温搅拌10min后,依次加入式2-1所示4-溴-3-三氟甲基苯胺(9.56g,40mmol)、式1-2环戊酮基双降冰片烯(nscsn)(4.81g,20mmol)继续搅拌若干分钟后,转移至150℃反应器中反应72h。待冷却至室温后,过滤除去不溶的无机盐,用乙酸乙酯淋洗,滤液浓缩后得粗产物。得到的粗产物通过快速柱层析提纯,得白色i-11二胺固体10.05g,产率90%。i-11为syn式与anti式两种位置异构体的混合物(即
ⅰ‑
syn-11和i-anti-11),
ⅰ‑
syn-11和
ⅰ‑
anti-11的摩尔比为1:1。
[0175]
上述二胺单体的合成路线如下:
[0176][0177]
所得产物的核磁氢谱参见图4,图4为实施例4中制备的二胺单体
ⅰ‑
syn-11和
ⅰ‑
anti-11的核磁氢谱图。
[0178]
2、制备聚酰亚胺薄膜
[0179]
在氩气氛围下,将所得二胺单体
ⅰ‑
11(5.58g,10.00mmol)溶于无水n,n-二甲基乙酰胺中,初始固含量为30wt%,室温搅拌至二胺单体完全溶解。然后在相同温度下加入式
ⅲ‑
4所示二酐单体(即s-bpda)(2.94g,10.00mmol),充分搅拌后,制得粘稠的聚酰胺酸溶液。在上述制备的聚酰酸溶液中加入吡啶(1.70ml,20.57mmol)和醋酸酐(2.00ml,21.02mmol)进行化学亚胺化。充分搅拌反应后,用甲醇沉淀出聚合物,随后将沉淀物重新溶解,倒入甲醇中再次析出,过滤,并重复2-3次后干燥得到聚酰亚胺共聚物。
[0180]
将所得聚酰亚胺共聚物溶解在间甲酚中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0181]
实施例5
[0182]
1、制备二胺单体
[0183]
向反应瓶中加入双乙腈氯化钯(51.89mg,0.20mmol)、三(邻甲基苯基)磷(121.75mg,0.40mmol)、磷酸钾(8.49g,40mmol)和无水n-甲基吡咯烷酮(40ml),室温搅拌10min后,依次加入式2-5所示4-氯-2-氟-5-三氟甲基苯胺(8.52g,40mmol)、式1-3所示类蒽醌结构的双降冰片烯(dmoha)(4.81g,20mmol)继续搅拌若干分钟后,转移至150℃反应器中反应72h。待冷却至室温后,过滤除去不溶的无机盐,用乙酸乙酯淋洗,滤液浓缩后得粗产物。得到的粗产物通过快速柱层析提纯,得白色i-21二胺固体10.10g,产率85%。
ⅰ‑
21为syn式与anti式两种位置异构体的混合物(即i-syn-21和
ⅰ‑
anti-21),i-syn-21和
ⅰ‑
anti-21的摩尔比为1∶1。
[0184]
上述二胺单体的合成路线如下:
[0185][0186]
所得产物的核磁氢谱参见图5,图5为实施例5中制备的二胺单体i-syn-21和i-anti-21的核磁氢谱图。
[0187]
2、制备聚酰亚胺薄膜
[0188]
在氩气氛围下,将所得i-21二胺固体(5.94g,10.00mmol)溶于无水n,n-二甲基乙酰胺中,初始固含量为30wt%,室温搅拌至二胺单体完全溶解。然后在相同温度下加入式
ⅲ‑
1所示二酐单体(即cbda)(1.96g,10.00mmol),充分搅拌后,制得粘稠的聚酰胺酸溶液。在上述制备的聚酰酸溶液中加入吡啶(1.70ml,20.57mmol)和醋酸酐(2.00ml,21.02mmol)进行化学亚胺化。充分搅拌反应后,用甲醇沉淀出聚合物,随后将沉淀物重新溶解,倒入甲醇中再次析出,过滤,并重复2-3次后干燥得到聚酰亚胺共聚物。
[0189]
将所得聚酰亚胺共聚物溶解在无水n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0190]
实施例6
[0191]
1、制备二胺单体
[0192]
向反应瓶中加入双三苯基磷二氯化钯(140.38mg,0.2mmol)、1,1'-联萘-2,2'-双二苯膦(249.07mg,0.4mmol)、氢氧化钠(1.60g,40mmol)和无水三氟甲苯(40ml),室温搅拌10min后,依次加入式2-2所示的4-氯-2-三氟甲基苯胺(7.80g,40mmol)、式1-4所示含苯并环丁烷结构的双降冰片烯(npnbd)(5.72g,20mmol)继续搅拌若干分钟后,转移至150℃反应器中反应72h。待冷却至室温后,过滤除去不溶的无机盐,用乙酸乙酯淋洗,滤液浓缩后得粗产物。得到的粗产物通过快速柱层析提纯,得白色i-25二胺固体9.72g,产率88%。
ⅰ‑
25为syn式与anti式两种位置异构体的混合物(即
ⅰ‑
syn-25和
ⅰ‑
anti-25),
ⅰ‑
syn-25和
ⅰ‑
anti-25的摩尔比为1∶1。
[0193]
所得产物的核磁氢谱参见图6,图6为实施例6中制备的二胺单体
ⅰ‑
syn-25和
ⅰ‑
anti-25的核磁氢谱图。
[0194]
上述二胺单体的合成路线如下:
[0195][0196]
2、制备聚酰亚胺薄膜
[0197]
在氩气氛围下,将所得二胺单体
ⅰ‑
25(5.52g,10.00mmol)溶于无水γ-丁内酯中,初始固含量为30wt%,室温搅拌至二胺单体完全溶解。然后在相同温度下加入式
ⅲ‑
5所示二酐单体(即6fda)(4.44g,10.00mmol),充分搅拌后,制得粘稠的聚酰胺酸溶液。在上述制备的聚酰酸溶液中加入吡啶(1.70ml,20.57mmol)和醋酸酐(2.00ml,21.02mmol)进行化学亚胺化。充分搅拌反应后,用甲醇沉淀出聚合物,随后将沉淀物重新溶解,倒入甲醇中再次析出,过滤,并重复2-3次后干燥得到聚酰亚胺共聚物。
[0198]
将所得聚酰亚胺共聚物溶解在无水n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0199]
所得聚酰亚胺共聚物的核磁氢谱参见图7,图7为实施例6中制备的聚酰亚胺共聚物的核磁氢谱图。
[0200]
对比例1:制备聚酰亚胺薄膜
[0201]
按照实施例1的制备过程进行,不同的是,不引入syn式和/或anti式的
ⅰ‑
1含氟二胺单体(即
ⅰ‑
syn-1和
ⅰ‑
anti-1),而是将其替换为等摩尔量的1,4-环己二胺单体。具体如下:
[0202]
在氩气氛围下,将1,4-环己二胺(1.14g,10.00mmol)溶于无水n,n-二甲基乙酰胺中,初始固含量为30wt%,室温搅拌至二胺单体完全溶解。然后在相同温度下加入式
ⅲ‑
6所示二酐单体(即6fcda)(4.58g,10.00mmol),充分搅拌后,制得粘稠的聚酰胺酸溶液。在上述制备的聚酰胺酸溶液中加入吡啶(1.70ml,20.57mmol)和醋酸酐(2.00ml,21.02mmol)进行化学亚胺化。充分搅拌反应后,用甲醇沉淀出聚合物,将沉淀物在索式提取器中用甲醇作溶剂,索提回流48h后干燥得到聚酰亚胺共聚物。
[0203]
将所得聚酰亚胺共聚物溶解在n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺
溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0204]
对比例2:制备聚酰亚胺薄膜
[0205]
按照实施例1的制备过程进行,不同的是,不引入syn式和/或anti式的
ⅰ‑
1含氟二胺单体(即
ⅰ‑
syn-1和
ⅰ‑
anti-1),而是将其替换为等摩尔量的4,4
’‑
二氨基-2,2
’‑
双三氟甲基联苯(tfmb/tfdb)。具体如下:
[0206]
在氩气氛围下,将4,4
’‑
二氨基-2,2
’‑
双三氟甲基联苯(tfmb/tfdb)(3.20g,10.00mmol)溶于无水n,n-二甲基乙酰胺中,初始固含量为30wt%,室温搅拌至二胺单体完全溶解。然后在相同温度下加入式
ⅲ‑
6所示二酐单体(即6fcda)(4.58g,10.00mmol),充分搅拌后,制得粘稠的聚酰胺酸溶液。在上述制备的聚酰胺酸溶液中加入吡啶(1.70ml,20.57mmol)和醋酸酐(2.00ml,21.02mmol)进行化学亚胺化。充分搅拌反应后,用甲醇沉淀出聚合物,将沉淀物在索式提取器中用甲醇作溶剂,索提回流48h后干燥得到聚酰亚胺共聚物。
[0207]
将所得聚酰亚胺共聚物溶解在n,n-二甲基乙酰胺中,固含量为8wt%,将聚酰亚胺溶液过滤后倾倒于干燥洁净的玻璃板上,在80℃的恒温箱中保持6h后,将所得的该聚酰亚胺湿膜从玻璃板上剥离,固定在金属框架上进行拉伸,之后放入真空干燥箱中200℃干燥,得到所述的无色透明聚酰亚胺薄膜。
[0208]
实施例7
[0209]
对实施例1~6及对比例1~2所得聚酰亚胺薄膜(厚度均为30mm)进行光学性能、耐热性能测试测试,结果参见表1。
[0210]
表1薄膜的光学性能和耐热性能
[0211]
薄膜样品tg(℃)t
5%
(℃)λ
cut-off
(nm)t
400
(%)yi e313[d65/10]cte(ppmk-1
)实施例1391503272860.9825实施例2454505273871.0921实施例3431466270871.0624实施例4481510268860.9921实施例5418465290881.1120实施例6466470267891.0621对比例1360418320783.6032对比例2378430332713.9035
[0212]
其中,实施例1~6所得聚酰亚胺薄膜的动态热机械分析(dma)曲线、失重(tga)曲线及可见光区光学透过性能分别如图8~10所示,图8为对实施例1~6所得聚酰亚胺薄膜的动态热机械分析(dma)曲线图,图9为实施例1~6所得聚酰亚胺薄膜的失重(tga)曲线图,图10为实施例1~6所得聚酰亚胺薄膜的可见光区光学透过性能示意图。
[0213]
由表1测试结果可以看出,与对比例1~2相比,本发明制备的含氟高透明聚酰亚胺薄膜具有优异的光学性能、良好的耐热性能和尺寸稳定性能;具体的,薄膜的在400nm处的透光率(即t
400
(%))>85%,截止波长λ
cut-off
在290nm以下;玻璃化转变温度tg高于390℃;热膨胀系数cte在25ppmk-1
以下;黄度指数yi在1.2以下。
[0214]
本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,包括最佳方式,并且也使得本领域的任何技术人员都能够实践本发明,包括制造和使用任何装置或系统,和实施任何结合的方法。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。本发明专利保护的范围通过权利要求来限定,并可包括本领域技术人员能够想到的其他实施例。如果这些其他实施例具有近似于权利要求文字表述的结构要素,或者如果它们包括与权利要求的文字表述无实质差异的等同结构要素,那么这些其他实施例也应包含在权利要求的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
