1.本发明涉及一种物质的测定方法,尤其涉及一种4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及其有关物质的测定方法。
背景技术:
2.罗沙司他(roxadustat),化学名化学名为n-[(4-羟基-1-甲基-7-苯氧基-3-异喹啉)羰基]甘氨酸,主要用于治疗慢性肾病和终末期肾病相关的贫血症。由美国菲布罗根公司研发,是一种低氧诱导因子脯氨酸羟化酶抑制剂(hif-phi),能够在氧分压正常的情况下抑制hif-脯氨酰羟化酶,稳定hif-α,促进内源性促红细胞生成素erythropoietin,epo生成及改善铁吸收利用,综合调控促红细胞生成。在中国所进行的治疗透析依赖(dd-ckd)和非透析依赖(ndd-ckd)的慢性肾病患者贫血状况的两项iii期临床试验结果均达到了主要有效终点。
[0003]
4-羟基-7-苯氧基异喹啉-3-甲酸甲酯作为罗沙司他的起始物料,其分子式为c
17h13
no4,分子量为295.08,结构式为:
[0004][0005]
该起始物料的分析检测对反应控制和收率提高有着重要的作用,同时也直接影响着终产品的质量,通过分析本品原料药的合成工艺路线,对杂质谱进行梳理,本品涉及以下5种杂质:
[0006]
杂质a:杂质b:杂质c:杂质d:和杂质e:
[0007]
以上杂质带入到原料药或制剂中,直接影响其质量、安全性、有效性,现有技术中未见针对4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及上述有关物质测定方法的报道。
技术实现要素:
[0008]
发明目的:本发明旨在提供一种稳定有效且操作简单的4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及其有关物质的测定方法,能够有效分离4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及多个杂质,且峰形良好并达到分离度的要求。
[0009]
技术方案:本发明所述的4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及其有关物质的测定方法,包括如下步骤:
[0010]
(1)配制供试品溶液、对照溶液、分离度溶液、系统适用性溶液;
[0011]
(2)采用反相液相色谱法,流动相a为体积百分比为0.08%~0.12%三氟乙酸水溶液,流动相b为乙腈,采用梯度洗脱,流动相b的起始体积百分比为20%~30%,采用高效液相色谱仪进行检测分析;
[0012]
所述有关物质包括:
[0013]
杂质a:杂质b:杂质c:杂质d:和杂质e:
[0014]
优选地,所述梯度洗脱程序为:0~30min,流动相a体积百分比由70%~80%降至5%~15%,流动相b的体积百分比由20%~30%升至85~95%;30~35min,流动相a的体积百分比为5%~15%,流动相b的体积百分比为85~95%。
[0015]
优选地,所述梯度洗脱程序为:0~30min,流动相a体积百分比由70%~80%降至5%~15%,流动相b的体积百分比由20%~30%升至85~95%;30~35min,流动相a的体积百分比为5%~15%,流动相b的体积百分比为85~95%;35~35.1min,流动相a体积百分比由5%~15%升至70%~80%,流动相b的体积百分比由85~95%降至20%~30%;35.1~45min,流动相a的体积百分比为70%~80%,流动相b的体积百分比为20%~30%。
[0016]
优选地,所述梯度洗脱程序为:0~30min,流动相a体积百分比由75%降至10%,流动相b的体积百分比由25%升至90%;30~35min,流动相a的体积百分比为10%,流动相b的体积百分比为90%;35~35.1min,流动相a体积百分比由10%升至75%,流动相b的体积百分比由90%降至25%;35.1~45min,流动相a的体积百分比为75%,流动相b的体积百分比为25%。
[0017]
优选地,所述液相检测采用的检测器为紫外吸收检测器,其检测波长为255~265nm和225~235nm。
[0018]
优选地,所述液相检测采用的色谱柱以十八烷基硅烷键合硅胶为填充剂,色谱柱柱温为30~40℃。
[0019]
优选地,所述流动相流速为0.8~1.2ml/min。
[0020]
有益效果:与现有技术相比,本发明具有如下显著优点:(1)所述检测方法稳定有效且操作简单,能够有效分离4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及多个杂质,样品成分保留能力强,分离度好;(2)灵敏度高,杂质含量测定结果准确,在本发明的色谱条件下能够准确检测相关已知杂质,且主峰与杂质峰能够达到有效分离;(3)通过方法学验证,检测方法准确可行,专属性、重复性和线性良好。
附图说明
[0021]
图1为实施例1中空白溶液(260nm)的色谱图;
[0022]
图2为实施例1中空白溶液(230nm)的色谱图;
[0023]
图3为实施例1中分离度溶液(260nm)的色谱图;
[0024]
图4为实施例1中分离度溶液(230nm)的色谱图;
[0025]
图5为实施例1中供试品溶液(260nm)的色谱图;
[0026]
图6为实施例1中供试品溶液(230nm)的色谱图。
具体实施方式
[0027]
下面结合附图对本发明的技术方案作进一步说明。
[0028]
实施例1
[0029]
分离度考察
[0030]
高效液相色谱条件:色谱柱为十八烷基硅烷键合硅胶柱,型号为agilent eclipse xdb-c18柱(规格为4.6mm
×
150mm,填料粒径为3.5μm),以0.1%三氟乙酸水溶液为流动相a,乙腈为流动相b,按照下表1进行梯度洗脱,流速为每分钟1.0ml;检测波长为260nm和230nm;柱温为35℃。
[0031]
表1梯度洗脱
[0032]
时间(分钟)流动相a(%)流动相b(%)0.0752530.0109035.0109035.1752545.07525
[0033]
溶液配制:
[0034]
供试品溶液:取4-羟基-7-苯氧基异喹啉-3-甲酸甲酯适量,精密称定,加稀释剂乙腈超声使溶解并稀释制成每1ml约含4-羟基-7-苯氧基异喹啉-3-甲酸甲酯0.5mg的溶液。
[0035]
对照溶液:精密量取供试品溶液1.0ml,用乙腈稀释制成每1ml约含4-羟基-7-苯氧基异喹啉-3-甲酸甲酯0.5μg的溶液。
[0036]
杂质对照品贮备液:分别取杂质a对照品、杂质b对照品、杂质c对照品、杂质d对照品、杂质e对照品适量,分别加乙腈溶解并稀释制成每1ml约杂质d10μg,其余各杂质5μg的溶液。
[0037]
分离度溶液:取4-羟基-7-苯氧基异喹啉-3-甲酸甲酯供试品适量,精密称定,加乙腈超声使溶解,另取上述各杂质对照品贮备液适量,制成每1ml约杂质d1μg,其余各杂质0.5μg,4-羟基-7-苯氧基异喹啉-3-甲酸甲酯0.5mg的混合溶液。
[0038]
系统适用性溶液:取4-羟基-7-苯氧基异喹啉-3-甲酸甲酯对照品适量,精密称定,加乙腈超声使溶解,另取上述各杂质对照品贮备液适量,制成每1ml约杂质d1μg,其余各杂质0.5μg,4-羟基-7-苯氧基异喹啉-3-甲酸甲酯0.5mg的混合溶液。
[0039]
空白溶液:乙腈
[0040]
精密量取空白溶液、分离度溶液、供试品溶液进行测定。图谱见图1、图2、图3、图4、
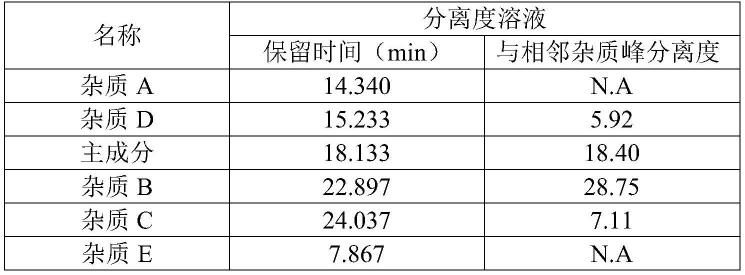
图5、图6,检测结果见表2。结果表明,空白溶液无干扰,主成分与杂质之间以及各杂质之间均能有效分离,表明本方法专属性良好。
[0041]
表2分离度检测结果
[0042][0043]
实施例2
[0044]
定量限、检测限考察
[0045]
参照实施例1中方法配制主成分(4-羟基-7-苯氧基异喹啉-3-甲酸甲酯)对照品和杂质对照品贮备液,分别精密量取各杂质对照品贮备液适量,用乙腈逐步稀释测定,将信噪比约为10:1时作为定量限,将信噪比约为3:1时作为检测限。并将定量限浓度的溶液,连续进样6次,考察定量限溶液进样精密度。结果见表3、表4。结果表明本方法灵敏度良好。
[0046]
表3检测限与定量限
[0047][0048]
表4定量限进样精密度试验结果
[0049]
s/n杂质a杂质d杂质b杂质c杂质e主成分125.339.317.812.113.635.3231.747.521.614.421.842.9330.146.120.514.816.141.7444.766.830.820.917.959.8525.139.118.011.114.635.4639.358.826.217.918.352.3均值32.749.622.515.217.144.6保留时间杂质a杂质d杂质b杂质c杂质e主成分114.09314.85022.63023.7677.69017.800214.09314.85022.63023.7777.69017.800314.09714.85022.63323.7707.67317.800
414.09714.85022.63023.7737.67317.800514.09314.85022.63023.7737.67317.797614.09714.85022.63323.7677.66717.803均值14.09514.85022.63123.7717.67717.800rsd(%)0.020.000.010.020.130.01峰面积杂质a杂质d杂质b杂质c杂质e主成分10.03080.04640.02410.01390.03390.047720.03160.04580.02470.01360.04170.046830.02920.04500.02240.01560.03720.046840.03240.04820.02420.01480.04280.045450.03110.04710.02500.01260.03840.049060.03210.04630.02500.01460.03650.0458均值0.03120.04650.02420.01420.03840.0469rsd(%)3.682.364.037.398.682.78
[0050]
实施例3
[0051]
线性考察
[0052]
参照实施例1中方法配制主成分(4-羟基-7-苯氧基异喹啉-3-甲酸甲酯)对照品和杂质对照品贮备液,分别精密量取各杂质对照品贮备液适量,用乙腈稀释制成一系列浓度,作为各成分的线性溶液。检测结果见下表5。采用标准曲线法测定校正因子,取线性结果测得的校正因子,结果见表6。结果表明,采用本发明的色谱条件,各待测成分线性关系良好。
[0053]
表5线性考察结果
[0054][0055]
表6校正因子
[0056]
名称斜率校正因子主成分-260nm0.4762/主成分-230nm0.4102/杂质a0.31721.5杂质b0.23792.0杂质c0.15553.1杂质d0.59610.80杂质e-230nm0.25431.6
[0057]
实施例4
[0058]
溶液稳定性考察
[0059]
分别于不同时间取系统适用性溶液、供试品溶液、对照溶液5μl注入液相色谱仪,记录色谱图。结果见表7、表8。结果表明,系统适用性溶液和对照溶液在室温条件下放置51小时,各杂质峰面积及主峰峰面积与0小时比较,比值均在90.0%~110.0%之间,供试品溶液在室温条件下放置51小时内,各时间点各杂质含量与0小时比较,均无大于报告限杂质检出。表明在室温条件下,系统适用性溶液、供试品溶液、对照溶液至少放置51小时内稳定。
[0060]
表7有关物质溶液稳定性试验-系统适用性溶液
[0061][0062]
表8有关物质溶液稳定性试验-对照溶液
[0063]
时间峰面积比值0h0.2630/7h0.257297.8%14h0.260899.2%21h0.2654100.9%28h0.2687102.2%36h0.2718103.3%44h0.2744104.3%51h0.2778105.6%
[0064]
实施例5
[0065]
重复性考察
[0066]
精密称取4-羟基-7-苯氧基异喹啉-3-甲酸甲酯样品6份,分别加入杂质限度100%的杂质对照品溶液,加乙腈溶解并稀释至刻度,作为供试品溶液。分别精密量取各供试品溶液稀释1000倍,作为对照溶液。分别精密量取上述供试品溶液和对照溶液5μl注入液相色谱仪,记录色谱图,以各杂质[(测的量-原有量)/加入量]计算回收率,结果见表9。结果表明,各杂质回收率均在80%~120%之间,rsd小于10.0%,表明本方法重复性良好。
[0067]
表9重复性计算表
[0068]
杂质名称平均回收率回收率rsd杂质a100.8%1.0%杂质b102.3%1.5%杂质c101.3%1.9%杂质d99.1%1.4%杂质e103.5%7.9%
[0069]
实施例6
[0070]
准确度考察
[0071]
精密称取4-羟基-7-苯氧基异喹啉-3-甲酸甲酯样品12份,分别加入杂质限度50%、100%、150%的杂质对照品溶液,加稀释剂溶解并稀释至刻度,作为供试品溶液。分别精密量取各供试品溶液稀释1000倍,作为对照溶液。分别精密量取上述供试品溶液和对照
溶液5μl注入液相色谱仪,记录色谱图,以各杂质[(测的量-原有量)/加入量]计算回收率,结果见表10。结果表明,各杂质回收率均在80%~120%之间,rsd小于10.0%,表明本方法准确度良好。
[0072]
表10准确度计算表
[0073]
杂质名称回收率范围平均回收率rsd杂质a99.80%~105.62%102.5%3.0%杂质b100.99%~109.20%104.2%2.2%杂质c95.44%~104.46%100.5%2.6%杂质d97.04%~103.38%100.1%3.0%杂质e90.74%~111.63%100.8%8.4%
[0074]
综上所述,本发明方法测定4-羟基-7-苯氧基异喹啉-3-甲酸甲酯及其有关物质,专属性强、灵敏度高、重复性和线性良好、准确度高。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。