1.本发明涉及电极材料制备技术领域,特别是一种点线面三合一高稳定性三维网络结构电极的制备方法。
背景技术:
2.硅负极因极高的理论比容量、适宜的工作电压、良好的安全性、环境友好等优点,被认为是理想的下一代负极材料。然而,其在循环过程中巨大的体积效应及低的电导率严重阻碍了其商业化应用。
3.通常来说:电极由活性材料、粘合剂和导电剂按一定比例混合组成,其中活性材料提供容量,粘结剂在于保持活性物质与集流体间的粘结作用,导电剂提供活性物质与集流体间的电接触。
4.为了解决硅负极的体积变化及导电性差的问题,现有技术中,针对活性材料和粘结剂方面的改性是提升电极稳定性的一般策略。例如,制备不同结构的(纳米管、多孔、石榴状结构等)硅负极已被证明是有效策略,通过减缓硅的体积膨胀来提高循环稳定性。粘结剂方面,包括自愈聚合物、软硬聚合物的组合以及导电弹性聚合物网络,在释放应力和提高粘结强度、稳定电极方面显示出明显的优势。然而,作为电极重要组成部分的导电剂却很少涉及,这是因为现有电极材料中,导电剂一般采用乙炔黑,或碳纳米管等,与活性物质及粘结剂间无相互键合作用力。因此尚无从导电剂的角度增强硅负极结构稳定性的相关研究或者报道。
5.公开号为cn113422058a的专利文献公开了一种mxene/snse基钾离子电池负极材料及其制备方法,其以snse为活性材料,通过引入mxene导电基体,使snse在mxene片层上原位负载,可以保证 snse的均匀分布,同时缓解snse在充放电过程中的体积膨胀,避免电极材料的粉碎剥落,所述mxene/snse作为钾离子电池负极材料时可提升其循环稳定性。然而,上述专利同样是针对于活性物质的改性,而非电极整体的稳定性提升。此外,该专利技术存在如下缺陷:
6.(1)从技术原理来看,该专利是使snse在mxene片层上进行原位负载,即该专利是针对活性材料进行改性来提高电极稳定性,是现有提升电极稳定性的常规方法,其在制备电极时需要将 mxene/snse复合材料、导电剂、粘结剂进行混合,即该专利所引入的mxene只是对活性材料进行改性,mxene本身并非作为导电剂使用,在形成电极时该专利同样需要额外添加导电剂;
7.(2)该专利是使snse在mxene片层上进行原位负载,但snse 与mxene直接的作用力较弱,在充放电循环过程中,随着活性材料的反复体积变化,活性材料会逐渐与mxene分离,这使得该专利技术并不能长期抑制活性材料的体积膨胀,也就是说通过原位负载使活性材料负载于mxene片层的技术方案,对抑制活性材料体积膨胀的影响有限;此外,该专利技术只能对snse这种特定的负极电极活性材料进行改性,对于其它诸如硅等负极材料甚至其它体积变化较大的阳极材料,该专利技术并不适用,即该专利技术的适用范围较低;
8.(3)该专利技术虽然合成了mxene/snse复合材料,但其在制备电极时同样需要与一定量的导电剂和粘结剂混合,以利用粘结剂将活性材料与导电剂粘结形成电极,这种物理混合的形式并未突破传统电极制备的限制,其活性材料、导电剂、粘结剂之间无法在微观结构层面进行键合以形成整体电极,而这种传统混合电极的稳定性的提升是受限的。
9.综上所述,为了提升电极的稳定性,现有技术都是从活性材料及粘结剂的角度出发对其进行改进,然而,作为电极重要组成部分的导电剂,它在硅和集流体之间提供了不可或缺的电接触,但现有技术中并未出现从导电剂角度出发来来提升电极稳定性的研究。比如炭黑 (cb)作为传统的导电剂,随着硅等活性材料的反复体积变化而容易团聚,从而导致内部硅失去电接触后成为死硅,进而导致电极循环性能恶化。此外,现有电极中的导电剂与活性材料及粘结剂之间没有键合作用,导电剂在稳定电极结构方面无任何作用。
10.前述内容所提到的公开号为cn113422058a的专利文献中所引入的mxene也仅是为了针对活性材料进行改性而非电极整体。而现有存在的以mxene为导电剂制备硅负极的技术,虽然其可以一定程度上抑制活性材料的体积膨胀但其仍然无法与活性物质产生有效的键合作用,因此但其容量衰减迅速,远远无法满足商业应用。。
技术实现要素:
11.为了克服现有技术的不足,本发明提供了一种点线面三合一高稳定性三维网络结构电极的制备方法,即本发明通过活性物质的表面改性及利用mxene作为导电剂,实现了导电剂与活性物质间的键合作用、导电剂与粘结剂间的交联作用,最终形成了点线面相结合的三维网络结构电极,大大提升了电极的整体稳定性及电化学性能。填充了从导电剂角度出发来提升电极稳定性的技术空白。
12.本发明解决其技术问题所采用的技术方案是:一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
13.s1.对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒;
14.s2.将经表面改性处理后的活性材料与mxene及粘结剂按一定比例在溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用mxene与活性材料间的键合作用、mxene与粘结剂间的交联作用形成点线面三维网络结构电极。
15.作为进一步的优选实施方案,所述电极活性材料为si、fe2o3, sno2、sn,ge,geo2中的一种。
16.作为进一步的优选实施方案,步骤s1中使用3-氨基丙基三乙氧基硅烷、3-氨丙基三甲氧基硅烷、聚二烯丙基二甲基氯化铵中的至少一种对电极活性材料进行表面改性。
17.作为进一步的优选实施方案,步骤s1中所述对电极活性材料进行表面改性的具体操作为:所述电极活性材料为硅,将100-1000mg 活性材料、10-100ml乙醇、1-5ml 3-氨基丙基三乙氧基硅烷及1-5ml 硝酸溶液混合,50-80℃下搅拌3-8h,然后抽滤并干燥处理。
18.作为进一步的优选实施方案,所述mxene为ti3c2tx、ti2ctx、 v2ctx中的至少一种。
19.作为进一步的优选实施方案,所述mxene的制备方法为:将 0.05-2g的ti3alc2合金粉末,加入到含10-50ml盐酸及1-5glif的溶液中,在35-50℃的水浴锅中连续搅拌24-48小时,离心并洗涤获得沉淀物,在氩气保护下通过超声处理沉淀物水溶液,获得ti3c2胶体悬浮液。
20.作为进一步的优选实施方案,步骤s2中所述溶剂为水。
21.作为进一步的优选实施方案,所述粘结剂为海藻酸钠、聚乙烯醇、羧甲基纤维素钠、聚丙烯酸中的至少一种。
22.作为进一步的优选实施方案,步骤s3中活性材料、mxene、粘结剂间的质量比为1~10:0.5~1.5:1~6。
23.作为进一步的优选实施方案,步骤s3中活性材料、mxene、粘结剂间的质量比为6:1:3。
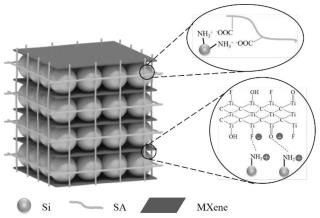
24.本发明的积极效果:本发明选用片状结构的mxene作为导电剂,同时利用mxene的终端基团与粘结剂基团之间的交联反应实现导电剂与粘结剂之间的强力结合,利用mxene与改性活性材料之间的偶极相互作用实现导电剂与活性材料间的强键合,最终mxene、改性活性材料、粘结剂三者合一形成具有三维网络结构的电极,其中点、线和面(片状物)分别代表活性材料、粘合剂和导电剂。本发明所制成的电极具有优异的电化学性能及较长的使用寿命,其中mxene作为导电剂,其不仅提供了良好的电接触,而且还有效地减轻了局部应力和缓冲了体积膨胀,同时由于其与活性材料和粘合剂的强结合作用,使电极稳定性得到极大提升。
25.传统电极的制备方式是将活性材料、导电剂、粘结剂进行简单混合,以通过粘结剂的粘结作用形成电极整体,其导电剂除了提供电接触作用更无其它用处,而本发明则突破了传统电极的制备方式,其导电剂与活性材料和粘结剂间均产生了强键合作用,三者合一后获得了在微观结构层面形成整体的三维网络结构电极,其相比于传统方法制备的电极具有更优异的电化学性能。
26.此外,本发明对于粘结剂及活性材料的选择也具有一定的灵活性,适用性强,为高能量密度锂离子电池电极的总体设计提供新的见解。
附图说明
27.图1为所述si-aps-m-sa电极的结构示意图;
28.图2a为实施例4所制备ti3c2tx的xrd图;
29.图2b为实施例4所制备ti3c2tx的tem图;
30.图2c为实施例4所制备ti3c2tx的afm图;
31.图2d为实施例4所制备ti3c2tx的厚度表征图;
32.图3为所述si-aps-m-sa的sem图;
33.图4为所述si-aps-m、所述si-aps-m-sa的xps对比图;
34.图5为不同电极的恒电流充放电测试结果图;
35.图6为不同电极的倍率性能测试结果图;
36.图7为所述si-aps-m-sa电极的长循环性能测试结果图;
37.图8a为循环测试前所述si-cb-sa的sem图;
38.图8b为循环测试前所述si-aps-cb-sa的sem图;
39.图8c为循环测试前所述si-m-sa的sem图;
40.图8d为循环测试前所述si-aps-m-sa的sem图;
41.图8e为循环测试后所述si-cb-sa的表面形貌图;
42.图8f为循环测试后所述si-aps-cb-sa的表面形貌图;
43.图8g为循环测试后所述si-m-sa的表面形貌图;
44.图8h为循环测试后所述si-aps-m-sa的表面形貌图;
45.图8i为循环测试前所述si-cb-sa的截面sem图;
46.图8j为循环测试前所述si-aps-cb-sa的截面sem图;
47.图8k为循环测试前所述si-m-sa的截面sem图;
48.图8l为循环测试前所述si-aps-m-sa的截面sem图;
49.图8m为循环测试后所述si-cb-sa的截面sem图;
50.图8n为循环测试后所述si-aps-cb-sa的截面sem图;
51.图8o为循环测试后所述si-m-sa的截面sem图;
52.图8p为循环测试后所述si-aps-m-sa的截面sem图;
53.图9为所述si-aps-m-pva电极的循环性能测试图;
54.图10为所述si-aps-m-cmc电极的循环性能测试图;
55.图11为所述sno
2-aps-m-sa电极的循环性能测试图;
56.图12为所述fe2o
3-aps-m-sa电极的循环性能测试图;
57.图13为全电池充放电循环图。
具体实施方式
58.下面结合附图对本发明的优选实施例进行详细说明。
59.实施例1
60.本发明实施例1提供一种改性硅电极活性材料,制备步骤如下:对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒,具体为:活性材料为si,将500mg si纳米颗粒、30ml无水乙醇、1.5ml 3-氨基丙基三乙氧基硅烷(aptes)及3ml硝酸溶液混合,60℃下搅拌5h,将产物进行抽滤处理,并分别用水和无水乙醇洗涤5次,干燥后得到aptes修饰后的si活性材料,记为siaps。
61.实施例2
62.本发明实施例2提供一种改性sno2电极活性材料,其制备步骤如下:
63.对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒,具体为:活性材料为sno2,将500mg sno2纳米颗粒、30ml无水乙醇、1.5ml 3-氨基丙基三乙氧基硅烷(aptes)及5ml蒸馏水混合,室温下搅拌5h,将产物进行抽滤处理,并分别用水和无水乙醇洗涤5次,干燥后得到aptes修饰后的sno2活性材料,记为sno
2 aps。
64.实施例3
65.本发明实施例3提供一种改性fe2o3电极活性材料,制备步骤如下:
66.对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒,具体为:活性材料为fe2o3,将500mg fe2o3纳米颗粒、2ml 3
‑ꢀ
氨基丙基三乙氧基硅烷(aptes)及100ml蒸馏水混合,室温下搅拌5h,将产物进行过滤,60℃下真空干燥12小时得到aptes修饰后的fe2o3活性材料,记为fe2o3aps。
67.实施例4
68.本发明实施例4提供一种mxene的制备方法,其步骤为:
69.将1g的ti3alc2合金粉末,缓慢加入到含10ml盐酸(12m)及 1.6glif的混合溶液中,在35-50℃的水浴锅中连续搅拌24-48小时,以剥离al层;离心数次,用去离子水洗涤沉淀物,直到上清液的ph 达到中性;然后,在氩气保护下通过超声处理2小时制备分层的ti3c2tx胶体溶液,以3500rpm转速离心0.5小时以去除沉积物后,获得多层ti3c2的ti3c2tx胶体悬浮液,ti3c2tx悬浮液的浓度为20 mg/ml,记为ti3c2tx溶液。
70.所制备获得的ti3c2t
x
纳米片,经x射线衍射(如图2a所示的 xrd图)和扫描电子显微镜(如图2b所示的sem图)证实了其具有典型的二维结构。使用原子力显微镜(如图2c所示的afm图)测量纳米片的厚度,厚度表征如图2d所示,通过测量可知获得了单层厚度为1.5nm的ti3c2t
x
纳米片,因为其巨大的比表面积,有助于与 si等活性材料结合,并提供良好的电流接触。
71.实施例5
72.本发明优选实施例5提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
73.将120mg实施例1制备的所述siaps、添加到1ml实施例4制备的ti3c2tx溶液中,超声20分钟后,经过滤干燥后获得的产物记为si-aps-m。
74.将120mg实施例1制备的所述siaps、添加到1ml实施例4制备的ti3c2tx溶液中,超声20分钟后,加入60mg海藻酸钠(sa) 与适量水,将溶液搅拌6小时,以彻底混合ti3c2tx、si aps和sa 形成浆料,其中siaps、ti3c2tx、海藻酸钠间的质量比为6:1:3;随后,用刮刀将均匀混合的浆料涂覆到铜箔上,然后在60℃下真空干燥12小时以去除水分,制备的电极记为si-aps-m-sa。
75.实施例6
76.本发明优选实施例6提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
77.将120mg实施例2制备的所述sno2aps、添加到0.25ml实施例 4制备的ti3c2tx溶液中,超声20分钟后,加入60mg海藻酸钠(sa) 与适量水,将溶液搅拌6小时,以彻底混合ti3c2tx、sno2aps和sa 形成浆料,其中sno2aps、ti3c2tx、海藻酸钠间的质量比为6:1:3;随后,用刮刀将均匀混合的浆料涂覆到铜箔上,然后在60℃下真空干燥12小时以去除水分,制备的电极记为sno
2-aps-m-sa。
78.实施例7
79.本发明优选实施例7提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
80.将120mg实施例3制备的所述fe2o
3 aps、添加到1ml实施例4 制备的ti3c2tx溶液中,超声20分钟后,加入60mg海藻酸钠(sa) 与适量水,将溶液搅拌6小时,以彻底混合ti3c2tx、fe2o
3 aps和 sa形成浆料,其中fe2o
3 aps、ti3c2tx、海藻酸钠间的质量比为6:1:3;随后,用刮刀将均匀混合的浆料涂覆到铜箔上,然后在60℃下真空干燥12小时以去除水分,制备的电极记为fe2o
3-aps-m-sa。
81.实施例8
82.本发明优选实施例8提供一种点线面三合一高稳定性三维网络结构电极的制备方法,制备方法与所述实施例5相同,不同的是将实施例5中的粘结剂进行替换,即将实施例5
中的海藻酸钠(sa)替换为聚乙烯醇(pva):制备的电极记为si-aps-m-pva。
83.实施例9
84.本发明优选实施例9提供一种点线面三合一高稳定性三维网络结构电极的制备方法,制备方法与所述实施例5相同,不同的是将实施例5中的粘结剂进行替换,即将实施例5中的海藻酸钠(sa)替换为羧甲基纤维素钠(cmc):制备的电极记为si-aps-m-cmc。
85.对比例1
86.本发明对比例1提供一种硅电极的制备方法,包括如下步骤:
87.将120mg si纳米颗粒、添加到1ml实施例4制备的ti3c2tx溶液中,超声20分钟后,加入60mg海藻酸钠(sa)与适量水,将溶液搅拌6小时,以彻底混合ti3c2tx、si和sa形成浆料,其中si、ti3c2tx、海藻酸钠间的质量比为6:1:3;随后,用刮刀将均匀混合的浆料涂覆到铜箔上,然后在60℃下真空干燥12小时以去除水分,制备的电极记为si-m-sa。
88.对比例2
89.本发明对比例2提供一种硅电极的制备方法,包括如下步骤:
90.将120mg si纳米颗粒、40mg炭黑(导电剂)、40mg海藻酸钠(sa) 与适量水混合均匀形成浆料,其中si、炭黑、海藻酸钠间的质量比为 6:1:3;随后,用刮刀将均匀混合的浆料涂覆到铜箔上,然后在60℃下真空干燥12小时以去除水分,制备的电极记为si-cb-sa。
91.对比例3
92.本发明对比例3提供一种硅电极的制备方法,包括如下步骤:
93.将120mg实施例1制备的所述siaps、40mg炭黑(导电剂)、 40mg海藻酸钠(sa)与适量水混合均匀形成浆料,其中siaps、炭黑、海藻酸钠间的质量比为6:1:3;随后,用刮刀将均匀混合的浆料涂覆到铜箔上,然后在60℃下真空干燥12小时以去除水分,制备的电极记为si-aps-cb-sa。
94.通常,ti3c2tx由于丰富的末端基团(-o,-f)而呈现负电性,而纳米硅由于其活性表面的-oh同样表现出负电性。在相同电学性质下,ti3c2tx和si之间除了可能的氢键外没有强键合作用,而氢键无法承受si的重复体积变化。经实验测试,未改性si和ti3c2tx的zeta 电位分别为:-25.1mv和-23.2mv,而经aptes处理后的si(即所述siaps)的电位为 13.1mv。显然,siaps和ti3c2tx之间可以发生静电相互作用,这比si与ti3c2tx之间可能存在的氢键作用强得多,且经aptes处理后,si的结构和形态均未被破坏。参照图3,由实施例5所获得的si-aps-m-sa的sem图像可知,ti3c2tx和sa紧紧包裹着siaps颗粒,这可以为活性材料提供良好的电接触,且极有利于缓解si的体积膨胀。
95.所获得的si-aps-m-sa电极结构模型如图1所示。对实施例5 中所获得的si-aps-m及si-aps-m-sa进行x射线光电子能谱(xps) 测试以进一步揭示三维网络的固有结合,如图4所示,si-aps-m的 n1s区具有两个峰,分别为初级(-nh2)和次级(-nh)胺功能。而 si-aps-m-sa的n1s光谱在399.6ev处显示出一个额外的峰,对应于叔胺(-nr-co-)。n-c峰的出现归因于添加sa后酰胺的形成,与 c1s区域一致。这些结果表明,通过ti3c2tx交联sa和siaps可以实现具有三维网络结构的稳定电极。
96.在以锂为对电极的半电池体系中,测试了不同电极的电化学性能。在1ag-1
电流密度下进行充放电循环,以评估长循环性能。如图 5所示,200次循环后,si-cb-sa、si-aps-cb-sa、si-m-sa、 si-aps-m-sa电极的容量分别为451.8、698.6、1247.1和2162.7mahg-1
,相
应的容量保持率分别为13.7%、21.2%、36.4%和72.7%。 si-cb-sa和si-aps-cb-sa电极均表现出快速容量衰减,而含 ti3c2tx的电极具有较高的容量和保留率,说明ti3c2tx纳米片在抑制si体积膨胀方面具有明显的优势。在前50个循环中,si-m-sa和 si-aps-m-sa电极均显示出良好的电化学性能。然而,随着循环的进行,si-m-sa中发生了快速容量衰减,这是由于si与ti3c2tx之间仅有弱键合作用,在重复的体积变化过程中,si逐渐从ti3c2tx中分离,从而使得ti3c2tx的作用减弱。而si-aps-m-sa由于ti3c2tx与siaps 之间的强键合而表现出优异的循环性能,其中的ti3c2tx能够长期抑制si的体积膨胀。很明显,si电极结构的稳定性可以通过siaps、 ti3c2tx和sa的组合来实现。
97.不同极片的倍率性能测试如图6所示,当电流密度为0.5ag-1
和 1ag-1
时,含有ti3c2tx的电极的容量逐渐增加。当电流密度上升到 5c和10c时,si-m-sa电极的容量迅速下降,而si-aps-m-sa电极则略有下降,表明si-aps-m-sa电极通过强键合形成了稳定的电极,即使在高电流密度下也保持了良好的电接触。当电流密度升至10ag-1
时,si-aps-cb-sa、si-cb-sa、si-m-sa、si-aps-m-sa电极容量分别为772.2、825.9、1117.4和1240.2mah g-1
。与含cb的电极相比,含ti3c2tx的电极具有较高的容量,表明si-apsm-m-sa电极通过强键合形成了稳定的电极,即使在高电流密度下也保持了良好的电接触。当电流密度恢复到1a g-1
时,si-cb-sa、si-aps-cb-sa、 si-aps-m-sa、si-m-sa电极的容量分别为2395.6、2519.5、3055.2 和3219.5mah g-1
,表明si-aps-m-sa电极通过ti3c2tx与siaps和 sa的交联键合而具有稳定的结构。
98.在0.1a g-1
下活化后,在电流密度为1a g-1
的条件下对 si-aps-m-sa电极进行长期循环性能测试。如图7所示,与初始容量 3122.8mah g-1
相比,500次循环后,si-aps-m-sa电极可以保持 1447.5mah g-1
的比容量,保留率为46.35%,电极每次循环仅衰减 0.15%,显示出优异的循环稳定性。
99.以ncm811为正极,si-aps-m-sa为负极组装全电池,如图13 所示,在30次循环后的能量密度为564.5wh kg-1
,远大于目前商业 licoo2//石墨电池的容量(335wh kg-1
)。
100.使用sem评估循环测试前后(1a g-1
下循环200次)电极的形态变化。如图8a~8d所示,循环之前,可以在si-m-sa和si-aps-m-sa 电极中看到明显的二维片状结构,而含cb的电极呈现粒状成分。如图8e~8h所示,si-cb-sa和si-aps-cb-sa电极在200次循环后出现了由材料剥落引起的明显开裂和空洞,而含有ti3c2tx的电极保持了良好和整洁的表面;此外,si-m-sa电极上产生了深而宽的裂纹,而si-aps-m-sa电极中只能看到短而窄的裂纹。通过电极横截面的变化,可以很容易地观察到电极的体积膨胀。如图8i-图8l所示,循环之前,电极的厚度几乎没有差异,但在1ag-1
的条件下进行200次循环后,电极厚度产生了明显的变化,如图8m-图8p所示,si-aps-m-sa电极的厚度增加了64.1%,而si-cb-sa、si-aps-cb-sa 和si-m-sa电极的厚度增加分别为257.3%、209.7%和169.3%,远高于si-aps-m-sa电极,这表明通过导电剂(mxene)与改性活性材料(siaps)和粘结剂(sa)之间强键合作用,电极的体积膨胀可以被大大抑制,这正是本发明技术方案的关键所在。
101.上述实施例8和实施例9分别以聚乙烯醇、羧甲基纤维素钠替代海藻酸钠为粘结剂制备了硅电极,如图9及图10所示,实施例8制成的si-aps-m-pva电极,100次循环后此电极显示出1935.5mah g-1
的容量,实施例9制成的si-aps-m-cmc电极,100次循环后此电极显示出2028.4mah g-1
的容量,两者均显示出良好的电化学性能,由此可见,不同粘结剂的使用为
本发明技术方案了更多的选择。
102.与此同时,除硅电极外,本发明技术方案还适用于其它体积变化较大的电极材料,如实施例6和实施例7分别制备了sno
2-aps-m-sa 和fe2o
3-aps-m-sa电极。由图11可知,sno
2-aps-m-sa电极经50 次循环后比容量为583.1mah g-1
,而传统sno
2-cb电极经50次循环后的容量为394.4mah g-1
;由图12可知,fe2o
3-aps-m-sa电极经50 次循环后比容量为948.5mah g-1
,是传统fe2o
3-cb电极经50次循环后容量(298.1mah g-1
)的三倍多。
103.总之,本发明的技术方案在粘结剂及活性材料的选择中具有一定的普适性。
104.此外,本发明还提供如下实施例:
105.实施例10
106.本发明优选实施例10提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
107.s1.对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒,具体为:活性材料为si,将100mg si纳米颗粒、10ml乙醇、 1ml 3-氨基丙基三乙氧基硅烷(aptes)及1.5ml硝酸溶液混合,50℃下搅拌3h,将产物进行抽滤处理,并分别用水和无水乙醇洗涤5 次,干燥后得到aptes修饰后的si活性材料;
108.s2.mxene的制备:将0.05g的ti3alc2合金粉末,缓慢加入到含 10ml盐酸(12m)及1.6g lif的混合溶液中,在35℃的水浴锅中连续搅拌24小时,以剥离al层;离心数次,用去离子水洗涤沉淀物,直到上清液的ph达到中性;然后,在氩气保护下通过超声处理2小时制备分层的ti3c2tx胶体溶液,以3500rpm转速离心0.5小时以去除沉积物后,获得了多层ti3c2tx的胶体悬浮液;
109.s3.将步骤s1中制备的活性材料、步骤s2制备的mxene、粘结剂(海藻酸钠)在水溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用mxene与活性材料间的键合作用、mxene 与粘结剂间的交联作用形成点线面三维网络结构电极,其中活性材料、mxene、粘结剂间的质量比为6:0.5:3.6。
110.实施例11
111.本发明优选实施例11提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
112.s1.对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒,具体为:活性材料为si,将1000mg si纳米颗粒、100ml乙醇、5ml 3-氨基丙基三乙氧基硅烷(aptes)及5ml硝酸溶液混合, 80℃下搅拌8h,将产物进行抽滤处理,并分别用水和无水乙醇洗涤5 次,干燥后得到aptes修饰后的si活性材料;
113.s2.mxene的制备:将2g的ti3alc2合金粉末,缓慢加入到含 50ml盐酸(12m)及5glif的混合溶液中,在50℃的水浴锅中连续搅拌48小时,以剥离al层;离心数次,用去离子水洗涤沉淀物,直到上清液的ph达到中性;然后,在氩气保护下通过超声处理2小时制备分层的ti3c2tx胶体溶液,以3500rpm转速离心0.5小时以去除沉积物后,获得了多层ti3c2tx的胶体悬浮液;
114.s3.将步骤s1中制备的活性材料、步骤s2制备的mxene、粘结剂(海藻酸钠)在水溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用mxene与活性材料间的键合作用、mxene 与粘结剂间的交联作用形成点线面三维网络结构电极,其中活性材料、
mxene、粘结剂间的质量比为3:1:1。
115.实施例12
116.本发明优选实施例12提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
117.s1.对电极活性材料进行表面改性,以获得带正电性的活性材料颗粒,具体为:活性材料为si,将300mg si纳米颗粒、70ml乙醇、 4ml 3-氨基丙基三乙氧基硅烷(aptes)及2ml硝酸溶液混合,65℃下搅拌6h,将产物进行抽滤处理,并分别用水和无水乙醇洗涤5次,干燥后得到aptes修饰后的si活性材料;
118.s2.mxene的制备:将1.5g的ti3alc2合金粉末,缓慢加入到含 20ml盐酸(12m)及3.5glif的混合溶液中,在45℃的水浴锅中连续搅拌35小时,以剥离al层;离心数次,用去离子水洗涤沉淀物,直到上清液的ph达到中性;然后,在氩气保护下通过超声处理2小时制备分层的ti3c2tx胶体溶液,以3500rpm转速离心0.5小时以去除沉积物后,获得了多层ti3c2tx的胶体悬浮液;
119.s3.将步骤s1中制备的活性材料、步骤s2制备的mxene、粘结剂(海藻酸钠)在水溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用mxene与活性材料间的键合作用、mxene 与粘结剂间的交联作用形成点线面三维网络结构电极,其中活性材料、mxene、粘结剂间的质量比为2:1.5:1。
120.实施例13
121.本发明优选实施例13提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
122.s1.对sn电极活性材料进行表面改性,以获得带正电性的sn活性材料颗粒;其中,使用3-氨丙基三甲氧基硅烷对电极活性材料进行表面改性。
123.s2.将经表面改性处理后的sn活性材料与现有ti2ctx (mxene)及聚丙烯酸按一定比例在溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用ti2ctx与改性sn活性材料间的键合作用、ti2ctx与粘结剂聚丙烯酸间的交联作用形成点线面三维网络结构电极,其中活性材料、mxene、粘结剂间的质量比为 5:1:4。
124.实施例14
125.本发明优选实施例14提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
126.s1.对ge电极活性材料进行表面改性,以获得带正电性的ge 活性材料颗粒;其中,使用聚二烯丙基二甲基氯化铵对电极活性材料进行表面改性。
127.s2.将经表面改性处理后的ge活性材料与现有v2ctx(mxene) 及聚丙烯酸按一定比例在溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用v2ctx与改性ge活性材料间的键合作用、v2ctx与粘结剂聚丙烯酸间的交联作用形成点线面三维网络结构电极,其中活性材料、mxene、粘结剂间的质量比为2:1:1。
128.实施例15
129.本发明优选实施例15提供一种点线面三合一高稳定性三维网络结构电极的制备方法,包括如下步骤:
130.s1.对geo2电极活性材料进行表面改性,以获得带正电性的 geo2活性材料颗粒;其
中,使用3-氨丙基三甲氧基硅烷对电极活性材料进行表面改性
131.s2.将经表面改性处理后的geo2活性材料与现有ti3c2tx (mxene)及海藻酸钠按一定比例在溶剂中混合均匀形成浆料,将浆料均匀涂覆在铜箔上并经干燥处理,利用ti3c2tx与改性geo2活性材料间的键合作用、ti3c2tx与粘结剂海藻酸钠间的交联作用形成点线面三维网络结构电极,其中活性材料、mxene、粘结剂间的质量比为6:1:3。
132.以上所述的仅为本发明的优选实施例,所应理解的是,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,并不用于限定本发明的保护范围,凡在本发明的思想和原则之内所做的任何修改、等同替换等等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
