一种m6a测序的建库方法
技术领域
1.本发明属于测序技术领域,具体地,涉及一种m6a测序的建库方法。
背景技术:
2.表观转录组是一种基于rna生物化学修饰的功能相关的转录组组学,它开启了研究基因表达的转录后调控之门。自从60年前在trna中发现假尿苷以来,迄今为止,已发现超过150种rna修饰。1970年首次被发现以来,m6a被认为是哺乳动物mrna中丰度最高的修饰之一。m6a在动物mrna中占比0.2%-0.6%。最近关于m6a定位、功能及分子机制的研究证明这种修饰是可逆动态的,参与调节rna周期的多个步骤,包括rna剪切、rna衰老、pre-mirna加工和蛋白翻译等过程。
3.mrna中m6a修饰的动态的变化已经被证明与干细胞分化、应激反应、dna损伤修复、性别分化和生物钟周期紧密相关。此外,最近的研究发现,m6a的失调在人类癌症的发生和发展过程中发挥着至关重要的作用。m6a相关的酶,包括writers、readers、erasers,在许多肿瘤样本中都表达异常。例如,由缺氧引起的m6a去甲基化酶alkbh5可以引起干细胞的marker基因nanog的去甲基化,进而导致nanog基因表达和乳腺癌干细胞的量增加。此外,alkbh5通过增强foxmm1基因的表达来维持胶质母细胞瘤增殖和肿瘤发生。类似地,m6a去甲基化酶fto和甲基化转移酶3(mettl3)的致瘤功能已经分别在急性骨髓性白血病和肺癌中得到证明。
4.merip-seq被开发用来在全转录组范围内识别m6a修饰。在人类细胞中接近7000个编码蛋白的基因中,已发现超过10,000个m6a位点。尽管最近这这项技术已经得以改进,并且商已有业化的试剂盒,但是仍然rna的起始量仍然需要200-300μg。因为临床样本可以获得的rna总量很少,这就使得m6a在临床样本中的研究受限。尽管m6a是动物mrna中丰度最高的修饰,但是由于缺乏有效的方法,m6a在人类疾病,尤其是癌症中的作用还有很多未知。
技术实现要素:
5.为了解决上述技术问题中的至少一个,本发明的技术方案如下:
6.本发明提供一种m6a测序的建库方法,包括以下步骤:
7.s1,获得目标生物片段化后的rna样本;
8.s2,取一部分片段化后的rna,与带有m6a抗体的磁珠共孵育,洗脱后得到带有m6a修饰的rna片段,与另取部分片段化后的rna继续下述步骤分别建立ip文库和input文库;
9.s3,将rna片段把转录成cdna,并进行第一pcr扩增,添加测序接头;
10.s4,将第一pcr扩增的产物进行磁珠纯化,并用zapr和r-probes去除核糖体cdna,
11.s4,将去除核糖体cdna的cdna进行第二pcr扩增,并将第二pcr扩增的产物进行磁珠纯化,得到ip文库和input文库。
12.在本发明的一些实施方案中,步骤s1中,获得目标生物的rna样本后,若rin=2-3,则不进行片段化,若rin=4-6,则将rna样本置于94℃3min进行片段化,若rin≥7,则将rna
样本置于94℃5min进行片段化。
13.在本发明的一些实施方案中,所述带有m6a抗体的磁珠的获取步骤如下:
14.(1)取磁珠放置在磁力架上,待溶液澄清后,吸弃上清。
15.(2)加入800μl c1 buffer,吹打混匀,放置在磁力架上,待溶液澄清后,吸弃上清,重复此步骤3次;
16.(3)取50μl无核酸水加入抗体,摇匀,静置2min;
17.(4)在磁珠管中加入450μl的c1 buffer与抗体,吹打混匀;再加入等体积的c2 buffer,吹打混匀;
18.(5)用封口膜包裹ep管,置于恒温震荡金属浴,37℃,1400rpm孵育16-24h;
19.(6)将ep管放置在磁力架上至少1min,待溶液澄清后,弃上清;
20.(7)加入800μl hb,吹打混匀,将ep管放置在磁力架上至少1min,待溶液澄清后,弃上清;
21.(8)加入800μl lb,吹打混匀,弃上清;
22.(9)加入800μl sb,吹打混匀,弃上清;
23.(10)重复步骤(9)1~3次;
24.(11)加入800μl sb,吹打混匀,置于垂直混匀仪上混匀15min。
25.在本发明的一些实施方案中,步骤s2中,所述共孵育的步骤具体为:
26.(1)向片段化后的rna中加入带有m6a抗体的磁珠;
27.(2)片段化后的rna与带有m6a抗体的磁珠混合液在室温下于垂直混匀仪上7圈/min,结合1~2小时;
28.(3)置于磁力架上5min以上,待溶液澄清,弃上清。
29.在本发明的一些实施方案中,共孵育后,进一步包括对带有m6a抗体的磁珠进行清洗的步骤:
30.(1)带有m6a抗体的磁珠加入300μl的m6a binding buffer,轻轻吹打混匀,室温下孵育5min,将样本置于磁力架,待溶液澄清后,弃上清;
31.(2)加入300μl low salt buffer,静置30s,弃上清;
32.(3)加入300μl high salt buffer,静置30s,弃上清;
33.(4)加入300μl tet buffer,静置30s,弃上清,
34.其中,各缓冲液的配方如下:
35.m6a binding buffer:25mm ph7.4 tris-hcl,200mm nacl,1% np-40,4mm edta;
36.low salt buffer:0.2
×
sspe,0.1m edta,0.1% tween-20;
37.high salt buffer:0.2
×
sspe,0.1m edta,0.1% tween-20,300mm nacl;
38.tet buffer:30mm ph8.0 tris-hcl,5mm edta(ph8.0),0.05% tween-20。
39.在本发明的一些实施方案中,所述洗脱的步骤如下:
40.(1)加入预热的elution buffer,缓慢吹打混匀,在42℃下孵育5min,将ep管置于磁力架上3min;
41.(2)取新的1.5ml ep管,加入400μl结合液和600μl的无水乙醇;
42.(3)吸取磁力架上的上清至上述新的1.5ml ep管;
43.(4)再重复步骤(1)-(3)一次,
44.其中,elution buffer配方为:0.2m dtt,0.150m nacl,0.3m ph7.5 tris-hcl,0.1m edta,0.10% sds。
45.本发明的有益效果
46.相对于现有技术,本发明的有益效果是:
47.本发明公开了一种低起始量m6a的测序方法,可以检测样本起始量低至3μg,实验过程简捷,文库纯度高,质量好,浓度足够高,满足上机测序和后续的相关的研究,为理解人类疾病尤其是癌症中m6a修饰机制及动态变化机理提供了坚实的基础。
附图说明
48.图1示出了本发明的方法建立的cdna文库的检测结果图。
49.图2示出了利用本发明的建库方法测序得到的peak分布图。
50.图3示出了利用本发明的建库方法测序得到的s1样本的motif结果预测图。
51.图4示出了利用本发明的建库方法测序得到的s2样本的motif结果预测图。
52.图5示出了利用常规建库方法对低起始量mrna建库的检测结果图。
53.图6示出了利用常规建库方法测序得到的peak分布图。
54.图7示出了利用常规建库方法测序得到的s1样本的motif结果预测图。
55.图8示出了利用常规建库方法测序得到的s2样本的motif结果预测图。
具体实施方式
56.除非另有说明、从上下文暗示或属于现有技术的惯例,否则本技术中所有的份数和百分比都基于重量,且所用的测试和表征方法都是与本技术的提交日期同步的。在适用的情况下,本技术中涉及的任何专利、专利申请或公开的内容全部结合于此作为参考,且其等价的同族专利也引入作为参考,特别这些文献所披露的关于本领域中的相关术语的定义。如果现有技术中披露的具体术语的定义与本技术中提供的任何定义不一致,则以本技术中提供的术语定义为准。
57.本技术中的数字范围是近似值,因此除非另有说明,否则其可包括范围以外的数值。数值范围包括以1个单位增加的从下限值到上限值的所有数值,条件是在任意较低值与任意较高值之间存在至少2个单位的间隔。对于包含小于1的数值或者包含大于1的分数(例如1.1,1.5等)的范围,则适当地将1个单位看作0.0001,0.001,0.01或者0.1。对于包含小于10(例如1到5)的个位数的范围,通常将1个单位看作0.1。这些仅仅是想要表达的内容的具体示例,并且所列举的最低值与最高值之间的数值的所有可能的组合都被认为清楚记载在本技术中。
58.术语“包含”,“包括”,“具有”以及它们的派生词不排除任何其它的组分、步骤或过程的存在,且与这些其它的组分、步骤或过程是否在本技术中披露无关。为消除任何疑问,除非明确说明,否则本技术中所有使用术语“包含”,“包括”,或“具有”的组合物可以包含任何附加的添加剂、辅料或化合物。相反,出来对操作性能所必要的那些,术语“基本上由
……
组成”将任何其他组分、步骤或过程排除在任何该术语下文叙述的范围之外。术语“由
……
组成”不包括未具体描述或列出的任何组分、步骤或过程。除非明确说明,否则术语“或”指列出的单独成员或其任何组合。
59.为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。
60.实施例
61.以下例子在此用于示范本发明的优选实施方案。本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。
62.除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。
63.那些本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。
64.下述实施例中未作具体说明的分子生物学实验方法,均按照《分子克隆实验指南》(第四版)(j.萨姆布鲁克、m.r.格林,2017)一书中所列的具体方法进行,或者按照试剂盒和产品说明书进行。其他实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
65.实施例1低起始量建库方法
66.1.免疫磁珠与m6a抗体预混
67.利用dynabeads抗体偶联试剂盒(dynabeads antibody coupling kit,invitrogen,14311d)进行。
68.提前半小时开启超净工作台紫外灯,将磁力架、移液器、耗材(镊子、黄板)、nf水等紫外灭菌30min以上,下列步骤(1)-(4)在超净台进行。
69.若第一次使用,可将所有磁珠(60mg)溶于1.2ml 100%dmso中,按体积比分装,分次取用(每管200μl,10mg,共6管,每管以12-16个样本为准)4℃保存。
70.提前15min取出抗体(sysy、50mg)和分装好的磁珠,室温静置10min,然后转移至超净台开始实验。
71.(1)取分装的磁珠放置在磁力架上1min,待溶液澄清后,吸弃上清。
72.(2)加入800μl c1 buffer,吹打混匀,放置在磁力架上1min,待溶液澄清后,吸弃上清。重复此步骤3次。
73.(3)同时取50μl无核酸水(nf水)加入抗体,摇匀,静置2min。
74.(4)在磁珠管中加入所需量体积(450μl)的c1 buffer与抗体(50μl),吹打混匀;再加入等体积的c2 buffer(500μl),吹打混匀,如表1所示:
75.表1各试剂用量
[0076][0077]
(5)用封口膜包裹ep管,置于恒温震荡金属浴,37℃,1400rpm孵育16-24h。note:在第一次使用hb与lb清洗液之前,加入0.01%-0.1%的tween-20(15μl)。
[0078]
(6)将ep管放置在磁力架上至少1min,待溶液澄清后,弃上清。
[0079]
(7)加入800μl hb(磁珠用量大于20mg时,体积加倍),吹打混匀,将ep管放置在磁力架上至少1min,待溶液澄清后,弃上清。
[0080]
(8)加入800μl lb(磁珠用量大于20mg时,体积加倍),吹打混匀,弃上清。
[0081]
(9)加入800μl sb(磁珠用量大于20mg时,体积加倍),吹打混匀,弃上清。
[0082]
(10)重复步骤(9)一次(根据抗体用量酌情增加重复次数)。
[0083]
(11)加入800μl sb(磁珠用量大于20mg时,体积加倍),吹打混匀,置于垂直混匀仪上混匀15min。
[0084]
2.取样及处理
[0085]
(1)取3μg动物(具体物种)的rna(补水到20μl),加入20μl 2
×
fragment,之后非情况进行处理:
[0086]
情况1(片段化):
[0087]
①
rin=4-6时,94℃3min;
[0088]
②
rin≥7时,94℃5min。
[0089]
情况2(不片段化):
[0090]
rin=2-3。
[0091]
(2)取4μl作为input,直接接一链反应。
[0092]
(3)配置ip反应液,如表2所示。
[0093]
表2 ip反应液配方
[0094]
组分用量(μl)ip样本36nf水1440.5m edta30总计210
[0095]
(4)将1中步骤(11)步的ep管放置在磁力架上至少1min,待溶液澄清后,吸弃上清。
[0096]
(5)加入92μl
×
样本数体积的sb,吹打混匀,若不立即使用,置于4℃冰箱待用(不要超过48h)。
[0097]
3.m6a-免疫磁珠与rna片段相结合
[0098]
(1)向上一步中已经片段化后的210μl rna加入90μl已经预混好的m6a-免疫磁珠,整个体系增至300μl。
[0099]
(2)将这些片段化后的rna与m6a-免疫磁珠在室温下于垂直混匀仪上7圈/min,结合1小时(不要超过2小时)。
[0100]
(3)将ep管放置在磁力架上至少5min,待溶液澄清,弃上清。
[0101]
4.m6a-免疫磁珠清洗
[0102]
各缓冲液的配方如下:
[0103]
m6a binding buffer:25mm tris-hcl(ph7.4),200mm nacl,1% np-40,4mm edta。
[0104]
low salt buffer:0.2
×
sspe,0.1m edta,0.1% tween-20。
[0105]
high salt buffer:0.2
×
sspe,0.1m edta,0.1% tween-20,300mm nacl。
[0106]
tet buffer:30mm tris-hcl(ph8.0),5mm edta(ph8.0),0.05% tween-20。
[0107]
elution buffer:0.2m dtt,0.150m nacl,0.3m tris-hcl(ph7.5),0.1m edta,0.10% sds。提前准备42℃水浴锅,预热elution buffer。
[0108]
(1)向3中步骤(3)中的免疫磁珠复合物中加入300μl的m6a binding buffer,轻轻吹打混匀,室温下孵育5min。将样本置于磁力架,待溶液澄清后,弃上清,之后所有步骤都在磁力架上操作。
[0109]
(2)加入300μl low salt buffer,静置30s,弃上清。
[0110]
(3)加入300μl high salt buffer,静置30s,弃上清。
[0111]
(4)加入300μl tet buffer,静置30s,弃上清。离心,再用10μl小枪头去除残留。
[0112]
5.m6a rna洗脱(将免疫磁珠上的rna片段洗脱下来)
[0113]
使用rna clean&concentrator-5(dnase not included)(zymo,r1016)进行rna的洗脱。
[0114]
(1)将4中的ep管从磁力架上取下,加入预热的100μl elution buffer,缓慢吹打混匀,在42℃下孵育5min,将ep管置于磁力架上3min。
[0115]
(2)取新的1.5ml ep管,加入400μl结合液和600μl的无水乙醇。
[0116]
(3)吸取磁力架上的上清至上述新的1.5ml ep管。
[0117]
(4)再重复步骤(1)-(3)一次,目的是将磁珠上rna片段完全洗脱。
[0118]
6.rna纯化
[0119]
使用rna clean&concentrator-5(dnase not included)(zymo,r1016)进行rna的纯化。
[0120]
(1)把上述混合液混匀。
[0121]
(2)取800μl混合液加入离心柱子,12000g离心1min,弃下清。
[0122]
(3)重复步骤(2)的操作直至所有液体过柱完成。
[0123]
(4)柱子上加入400μl预洗液,12000g离心1min,弃下清。
[0124]
(5)柱子上加入700μl洗涤液,12000g离心1min,弃下清。
[0125]
(6)柱子上加入400μl洗涤液,12000g离心1min,弃下清。
[0126]
(7)空离12000g,2min。
[0127]
(8)将柱子放入一新的1.5ml离心管,加10μl nuclease-free water,室温静置2min,12000g离心1min。
[0128]
(9)冰上保存,准备进行文库构建。
[0129]
实施例2文库构建
[0130]
1.一链cdna合成
[0131]
(1)按表3配置混合液:
[0132]
表3混合液配置
[0133][0134]
将混合液置于pcr仪上,运行如下程序:
[0135]
72℃ 3min;4℃ 2min。
[0136]
(2)按表4配置一链合成母液:
[0137]
表4一链合成母液(first strand master mix)
[0138][0139]
将5.5μl一链合成母液加入到(1)中混合液中,总体系10μl。置于pcr仪上,运行如下程序:
[0140]
42℃ 90min;70℃ 10min;4℃ forever(可以过夜)。
[0141]
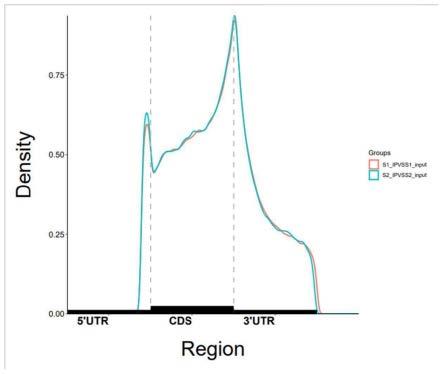
2.pcr1——加illumina接头和标签
[0142]
(1)按照表5准备pcr1反应体系(25μl):
[0143]
表5 pcr1反应体系
[0144][0145]
将上述14μl体系加到上一步反应液中再加入0.5μl forward primer,0.5μl reverse primer,置于pcr仪上运行下面程序:
[0146]
94℃ 1min;98℃ 15sec,55℃ 15sec,68℃ 30sec,5个循环;68℃ 2min;4℃ forever。
[0147]
3.文库纯化
[0148]
(1)将20μl纯化珠加入25μl pcr反应液中。用移液器吹打混匀。在室温下孵育8min,让cdna与磁珠结合。
[0149]
(2)将样品放在磁力架上5min或更长时间,直到液体完全澄清。
[0150]
(3)将样品在磁力架上,弃上清。
[0151]
(4)将样品放在在磁力架上;用200μl新鲜制成的80%乙醇添加到每个样品中。静置30sec,小心吸弃上清。
[0152]
(5)重复(4)一次。
[0153]
(6)让磁珠在室温下干燥约3min。
[0154]
(7)磁珠干燥后,加入26μl nf水并混匀。
[0155]
(8)在室温下孵育5min。
[0156]
(9)将样品放回磁力架上5min。
[0157]
(10)转移25μl透明上清液到无核酸酶的pcr管中。
[0158]
(11)重复步骤(1)~(6)进行第二次磁珠纯化。
[0159]
(12)磁珠干燥后,加入11μl zapr-master-mix去除核糖体cdna,从磁力架上取下pcr管,吹打混匀。
[0160]
(13)在室温下孵育5min以补水,短暂离心。
[0161]
(14)将样品放回磁力架上3min。
[0162]
(15)将10μl透明上清液转移到无核酸酶、低粘附性pcr管中。
[0163]
其中,步骤(12)中,利用zapr和r-probes去除核糖体cdna的具体步骤如下:
[0164]
将足以满足所需反应的r-probes等分试样移入pcr管中(每个反应0.75μl)。在pcr仪中进行以下程序:72℃ 2min,4℃保持。将r-probes管留在4℃的pcr仪中至少2min,但不超过10min。
[0165]
按表6准备zapr母液(zapr master mix):
[0166]
表6 zapr母液
[0167][0168][0169]
加入11μl zapr-master-mix至磁珠纯化管中,继续至(15)步。之后在pcr仪上运行以下程序:
[0170]
37℃ 60min,72℃ 10min,4℃保持。
[0171]
4.pcr2——最终rna测序文库扩增
[0172]
(1)按表7准备pcr2扩增体系:
[0173]
表7 pcr2扩增体系
[0174]
组分用量(μl)nf水132
×
seqamp pcr buffer25pcr2 primer mix1seqamp dna polymerase1
总计40
[0175]
其中,pcr2 primer mix包括两条通用引物,分别与模板上的p5/p7结合。
[0176]
将上述体系置于pcr仪上运行下面程序:
[0177]
94℃ 1min;98℃ 15sec,55℃ 15sec,68℃ 30sec,12个循环;68℃ 2min;4℃ forever。
[0178]
5.文库纯化
[0179]
(1)将50μl纯化珠加入50μl pcr2反应液中。用移液器吹打混匀。在室温下孵育5min,让cdna与磁珠结合。
[0180]
(2)将样品放在磁力架上5min,直到液体完全澄清。
[0181]
(3)将样品在磁力架上,弃上清。
[0182]
(4)将样品放在在磁力架上;用200μl新鲜制成的80%乙醇添加到每个样品中。静置30sec,小心吸弃上清。
[0183]
(5)重复(4)一次。
[0184]
(6)让磁珠在室温下干燥约3min。
[0185]
(7)磁珠干燥后,加入43μl nf水并混匀。
[0186]
(8)在室温下孵育5min。
[0187]
(9)将样品放回磁力架上5min。
[0188]
(10)将40μl透明上清液转移到无核酸酶、低粘附性pcr管中,保存到-20℃。
[0189]
nanodrop测量和bioanalyzer高灵敏度dna芯片。成功的cdna合成和扩增应产生一条跨越200
–
1,000bp的曲线,局部最大值在~300
–
400bp。本实施建库的检测结果如图1所示,可见cdna合成成功。
[0190]
实施例3测序与分析
[0191]
1.测序及数据处理
[0192]
测序产生的下机数据进行预处理,过滤掉不合格的序列后得到有效数据clean data,再进行下一步分析,具体处理步骤如下:
[0193]
(1)去除带接头的reads;
[0194]
(2)去除含有n(n表示无法确定碱基信息)的比例大于5%的reads;
[0195]
(3)去除低质量reads(质量值q≤10的碱基数占整个read的20%以上);
[0196]
(4)统计原始测序量,有效测序量,q20,q30,gc含量,如表8所示。
[0197]
表8测序数据
[0198]
sample_idraw_readsraw_basesvalid_readsvalid_basesvalid%q20%q30%gc%s1_ip375714405.64g369568965.25g93.0897.6293.2349.14s2_ip354859845.32g349975765.01g94.1797.8893.8249.47s1_input404827466.07g399547785.60g92.298.2894.8654.59s2_input404155246.06g397997785.69g93.8998.1994.7254.11
[0199]
注:
[0200]
sample_id:样本名;
[0201]
raw_bases:原始下机数据的碱基数;
[0202]
raw_reads:原始下机数据的reads数;
[0203]
valid_reads:有效数据的reads数;
[0204]
valid_bases:有效数据的reads数;
[0205]
valid%:有效reads所占比例;
[0206]
q20%:质量值≥20的碱基所占比例(测序错误率小于0.01);
[0207]
q30%:质量值≥30的碱基所占比例(测序错误率小于0.001);
[0208]
gc%:gc含量所占的比例。
[0209]
2.参考基因组比对
[0210]
使用hisat2对预处理后的有效数据(valid_reads)进行参考基因组比对,同时根据基因组注释文件gtf指定的基因位置信息分别统计,包括:(1)测序数据与参考基因组比对reads统计;(2)测序数据与参考基因组比对的染色体密度分布。比对结果如表9所示:
[0211]
表9比对结果
[0212][0213]
注:
[0214]
sample:样本名;
[0215]
valid reads:有效数据的reads数,即clean reads;
[0216]
mapped reads:能比对到基因组上的reads数;
[0217]
unique mapped reads:只能唯一比对到基因组某个位置的reads数;
[0218]
multi mapped reads:能比对到基因组多个位置的reads数;
[0219]
pe mapped reads:双端比对到基因组的reads数;
[0220]
reads map to sense strand:read比对到基因组上正义链的统计数;
[0221]
reads map to antisense strand:read比对到基因组上负义链的统计数;
[0222]
non-splice reads:read能够end-to-end比对到基因组区域,为整段比对统计;
[0223]
splice reads:read无法end-to-end比对到基因组区域,为分段比对统计。
[0224]
3.m6a甲基化富集区域寻找(peaking calling)
[0225]
发明人使用r包exomepeak2在全基因范围进行peak扫描,得到peak在基因组上的位置信息及peak长度信息等。结果如图2所示,发现了接近12,000个m6a峰,这些峰具有高的信噪比,将基因分为5’utr,cds,3’utr三个区段,统计在每个区段上peak的分布情况,peak一般富集于3’utr和stop codon区域。
[0226]
4.motif分析
[0227]
rna甲基化修饰以及去甲基化修饰起始于多种结合蛋白与发生甲基化位点的motif相结合。motif本质上是一种具有生物意义的核酸序列模式,这些rna甲基化相关的酶可识别这些motif并与之结合,从而影响基因的表达。基因表达调控机制研究是生物学研究的重点内容,鉴定这些motif,对于基因表达调控机制研究具有重要意义。使用motif分析软件homer在peak区域寻找可信度较高的motif,得到每个motif的宽度、p-value、%of targets、%of background等信息。我们默认针对每组样本进行motif预测。得到的结果如下:s1(图3)和s2(图4)样本均具有典型的rrach结构。
[0228]
实施例4常规建库方法m6a测序
[0229]
本实施例所述的常规建库方法是指采用universal low input rna kit for sequencing试剂盒进行m6a测序建库。
[0230]
若用3μg rna进行建库,发现无法达到要求,如图5所示。发明人采用200μg rna进行建库,测序结果如表10所示:
[0231]
表10常规建库测序数据
[0232]
sample_idraw_readsraw_basesvalid_readsvalid_basesvalid%q20%q30%gc%control_1_ip362167505.43g352685264.95g91.1097.9093.8349.42control_2_ip397124365.96g387380525.43g91.1997.9093.8349.42control_1_input459950746.90g453523386.33g91.7598.0494.1247.10control_2_input475393287.13g468642706.54g91.7098.0494.1047.14
[0233]
注:
[0234]
sample_id:样本名;
[0235]
raw_bases:原始下机数据的碱基数;
[0236]
raw_reads:原始下机数据的reads数;
[0237]
valid_reads:有效数据的reads数;
[0238]
valid_bases:有效数据的reads数;
[0239]
valid%:有效reads所占比例;
[0240]
q20%:质量值≥20的碱基所占比例(测序错误率小于0.01);
[0241]
q30%:质量值≥30的碱基所占比例(测序错误率小于0.001);
[0242]
gc%:gc含量所占的比例。
[0243]
与参考基因组比对结果如表11所示:
[0244]
表11常规建库测序结果与参考基因组比对
[0245][0246][0247]
注:
[0248]
sample:样本名;
[0249]
valid reads:有效数据的reads数,即clean reads;
[0250]
mapped reads:能比对到基因组上的reads数;
[0251]
unique mapped reads:只能唯一比对到基因组某个位置的reads数;
[0252]
multi mapped reads:能比对到基因组多个位置的reads数;
[0253]
pe mapped reads:双端比对到基因组的reads数;
[0254]
reads map to sense strand:read比对到基因组上正义链的统计数;
[0255]
reads map to antisense strand:read比对到基因组上负义链的统计数;
[0256]
non-splice reads:read能够end-to-end比对到基因组区域,为整段比对统计;
[0257]
splice reads:read无法end-to-end比对到基因组区域,为分段比对统计。
[0258]
m6a甲基化富集区域寻找结果如图6所示,motif预测如图7和图8所示。
[0259]
将基因分为5’utr,cds,3’utr三个区段,统计在每个区段上peak的分布情况,如图6所示,peak一般富集于3’utr和stop codon区域,具体分布情况也受物种,基因组注释,实验处理的影响。
[0260]
rna甲基化修饰以及去甲基化修饰起始于多种结合蛋白与发生甲基化位点的motif相结合。motif本质上是一种具有生物意义的核酸序列模式,这些rna甲基化相关的酶可识别这些motif并与之结合,从而影响基因的表达。基因表达调控机制研究是生物学研究的重点内容,鉴定这些motif,对于基因表达调控机制研究具有重要意义。使用motif分析软件homer在peak区域寻找可信度较高的motif,得到每个motif的宽度、p-value、%of targets、%of background等信息。发明人默认针对每组样本进行motif预测,结果如图7所示。
[0261]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。