llzo石榴石的氟化方法
1.本技术要求于2020年4月29日在欧洲以nr 20315228.5提交的优先权,出于所有目的将此申请的全部内容通过援引并入本技术。本技术涉及一种llzo石榴石的氟化方法。本发明还涉及通过所述方法获得的氟化无机化合物以及所述化合物作为锂电池的固体电解质的用途。
技术领域
2.石榴石型氧化物具有化学式a3b2(xo4)3的理想结构,并且通常上结晶成属于3-空间群的体心立方晶格。阳离子位点a、b和x分别具有viii、vi和iv的与氧的配位数。
3.合成石榴石主要以其磁性和介电特性而为人所知。然而,已经观察到某些石榴石可能具有足够高的li
离子电导率以将它们用作锂电池的固体电解质。因此,在2007年,团队成功地制备了具有式li7la3zr2o
12
的新型石榴石(llzo),并且获得了3
×
10-4
s/cm量级的总电导率。其他研究还表明,当石榴石具有立方结构而不是四方结构时,离子电导率最高。其他团队已经表明,当llzo石榴石包含另一化学元素(如铝或铌)时,离子电导率改善。
4.由于它们的高电导率,可以将llzo石榴石用作锂电池中的固体电解质。
背景技术:
5.ep 2353203 b1描述了通过共沉淀技术制备石榴石的方法。
6.wo 2019/090360描述了使llzo石榴石与锂盐(如lipf6或libf4)的溶液接触的方法。观察到图5中给出的nmr光谱与用本发明的产物获得的不同。
7.技术问题
8.llzo石榴石的表面能够在与大气中存在的水分和co2接触时被改性,这导致固体界面处的电导率的改变。这已经在例如phys.chem.chem.phys.[物理化学化学物理]2014,16(34),18294
–
18300https://doi.org/10.1039/c4cp02921f或j.mater.chem.a[材料化学杂志a]2014,2(1),172
–
181.https://doi.org/10.1039/c3ta13999a中得到了证明。具体地,观察到当石榴石颗粒与环境气氛接触时,在这些颗粒的表面处形成lioh和/或碳酸锂(在这方面还参见sharafi和sakamoto,j.mater.chem.[材料化学杂志]a,2017,5,13475)。
[0009]
因此,具有足够的离子电导率以用作锂电池的固体电解质并且可以在正常条件下储存和处理的石榴石将是有用的。
[0010]
本发明的方法旨在稳定所述石榴石而不降低其物理化学特性并且特别是其离子电导率。
附图说明
[0011]
图1表示在实例(即对比实例1)中用作起始材料的llzo型无机化合物m的ir-atr光谱。图2表示实例2的氟化无机化合物的ir-atr光谱。这两个光谱表示作为以cm-1
为单位的波数的函数的以任意单位(au)表示的信号强度。
[0012]
图3表示根据实例1制备的氟化llzo固体颗粒的sem-eds分析,即作为线轮廓上位
置的函数的所测量的元素f(k线)和la(m线)的绝对强度。
[0013]
图4表示根据对比实例2制备的氟化llzo固体颗粒(通过固态合成的氟化llzo)的sem-eds分析,即作为线轮廓上位置的函数的所测量的元素f(k线)和la(m线)的绝对强度。
技术实现要素:
[0014]
本发明的方法描述于权利要求1至11中。更准确地,该方法是一种氟化方法,其包括使包含二氟气体的气氛与具有石榴石型结构的无机化合物m接触,该无机化合物基于元素li、la、zr、a和o,并且对于该无机化合物,li、la、zr和a阳离子的相对组成对应于式(i):
[0015]
li
x
la3zr
zaw
ꢀꢀ(i)[0016]
其中:
[0017]
■
a表示选自由al、ga、nb、fe、w和ta形成的组的至少一种元素;
[0018]
■
x、z和w表示实数;
[0019]
■
1.20《z≤2.10;更特别地,1.20《z≤2.05;还更特别地,1.50≤z≤2.00;
[0020]
■
0《w≤0.80;更特别地,0《w≤0.60;还更特别地,0《w≤0.30;还更特别地,0《w≤0.25;
[0021]
■
4.00≤x≤10.50;更特别地,5.10≤x≤9.10;还更特别地,6.20≤x≤7.70。
[0022]
包含二氟气体的气氛由表述“氟化气氛”表示。
[0023]
本发明还涉及一种氧化物的氟化方法,该方法包括使含有二氟气体的气氛与具有式(ii)的氧化物接触:
[0024]
[li
x1
la3zr
zawo12
]
ꢀꢀ
(ii)
[0025]
其中:
[0026]
■
a表示选自由al、ga、nb、fe、w和ta形成的组的至少一种元素;
[0027]
■
x1、z和w表示实数;
[0028]
■
1.20《z≤2.10;更特别地,1.20《z≤2.05;还更特别地,1.50≤z≤2.00;
[0029]
■
0《w≤0.80;更特别地,0《w≤0.60;还更特别地,0《w≤0.30;还更特别地,0《w≤0.25;
[0030]
■
x1是正实数,其使得确保该氧化物的电中性。
[0031]
本发明还涉及通过本发明的方法获得的氟化无机化合物。该无机化合物是如权利要求12至26之一所定义的。
[0032]
本发明还涉及一种如权利要求27所定义的电解质,并且涉及如权利要求28和29所定义的氟化无机化合物的用途。
[0033]
现在将更详细地描述本发明。
具体实施方式
[0034]
起始无机化合物m具有石榴石型结构并且基于元素li、la、zr、a和o,对于该无机化合物,li、la、zr和a阳离子的相对组成对应于式(i):
[0035]
li
x
la3zr
zaw
ꢀꢀ(i)[0036]
其中:
[0037]
■
a表示选自由al、ga、nb、fe、w和ta形成的组的至少一种元素;
[0038]
■
x、z和w表示实数;
[0039]
■
1.20《z≤2.10;更特别地,1.20《z≤2.05;还更特别地,1.50≤z≤2.00;
[0040]
■
0《w≤0.80;更特别地,0《w≤0.60;还更特别地,0《w≤0.30;还更特别地,0《w≤0.25;
[0041]
■
4.00≤x≤10.50;更特别地,5.10≤x≤9.10;还更特别地,6.20≤x≤7.70。
[0042]
无机化合物m是基于元素li、la、zr、a和o的石榴石。由于元素铪通常天然存在于提取出锆的矿石中,并且因此存在于用于制备无机化合物m的起始化合物中,考虑到元素锆被元素铪部分替代,本技术中描述的所有内容也适用。因此,本发明还更特别地适用于包含元素铪的无机化合物m。因此,本发明可以更特别地适用于以基于元素li、la、zr、hf、a和o的石榴石形式的起始无机化合物m,对于该无机化合物,li、la、zr、hf和a阳离子的相对组成对应于式(ia):
[0043]
li
x
la3(zr
(1-a)
hfa)
zaw
ꢀꢀ
(ia)
[0044]
其中x、z和w如上所述,并且a是在0与0.05之间、更特别地在0与0.03之间、或甚至在0与0.02之间的实数。
[0045]
原子比hf/zr=a/(1-a)是在0与0.05之间、更特别地在0与0.03之间、或甚至在0与0.02之间。该比率可以在0.0006与0.03之间、或甚至在0.0006与0.025之间。
[0046]
a表示选自由al、ga、nb、fe、w和ta形成的组的至少一种元素或所述元素的组合。根据具体的实施例,a因此可以表示元素al与选自由ga、nb、fe、w和ta形成的组的元素a的组合。
[0047]
无机化合物m是电中性的。确保无机化合物m电中性的阴离子基本上是o
2-阴离子。然而,其他阴离子(例如像oh-和/或co
32-阴离子)也可能有助于无机化合物m的电中性。
[0048]
z可以在以下范围之一内:1.20《z≤2.10;更特别地,1.20《z≤2.05;还更特别地,1.50≤z≤2.00。更特别地,1.90≤z≤2.10。还更特别地,z≤2.00。
[0049]
w可以在以下范围之一内:0《w≤0.80;更特别地,0《w≤0.60;还更特别地,0《w≤0.30;还更特别地,0《w≤0.25。还更特别地,w≥0.05。
[0050]
阳离子的相对组成可以更特别地如下:
[0051]
■
a选自由nb、ta或这两种元素的组合形成的组;
[0052]
■
1.20《z≤2.10;更特别地,1.20《z≤2.05;更特别地,1.50≤z≤2.00;
[0053]
■
0.10《w≤0.80;更特别地,0.20《w≤0.80;更特别地,0.20《w≤0.50;
[0054]
■
6.20≤x≤10.35;更特别地,6.20≤x≤8.84;更特别地,6.50≤x≤7.48。
[0055]
阳离子的相对组成可以更特别地如下:
[0056]
■
a表示w;
[0057]
■
1.20《z≤2.10;更特别地,1.20《z≤2.05;更特别地,1.50≤z≤2.00;
[0058]
■
0.10《w≤0.80;更特别地,0.20《w≤0.80;更特别地,0.20《w≤0.50;
[0059]
■
5.40≤x≤10.20;更特别地,5.40≤x≤8.58;更特别地,6.00≤x≤7.26。
[0060]
阳离子的相对组成可以更特别地如下:
[0061]
■
a选自由al、ga、fe或这些元素的组合形成的组;
[0062]
■
1.90《z≤2.10;更特别地,1.95≤z≤2.05;更特别地,1.95≤z≤2.00;
[0063]
■
0.10《w≤0.80;更特别地,0.20《w≤0.60;更特别地,0.10《w≤0.25;
[0064]
■
4.60≤x≤10.05;更特别地,5.20≤x≤8.32;更特别地,6.25≤x≤7.37。
[0065]
无机化合物的经验式以及因此实数z、w和x的值是从无机化合物的化学分析推导出来的。为此,可以使用本领域技术人员已知的化学分析技术。这种方法可以包括制备由无机化合物m的化学侵蚀产生的溶液,并且然后确定该溶液的组成。例如,可以使用icp(电感耦合等离子体)、更特别地icp-ms(icp与质谱联用)或icp-aes(icp与原子发射光谱联用)。
[0066]
无机化合物m具有石榴石型结构。认为其结晶结构通常由lao8十二面体(la的配位数为8)的骨架和zro6八面体(zr的配位数为6)的骨架组成。更特别地,该结晶结构可以由配位数为8(24c位点)的lao8十二面体的骨架和配位数为6(16a位点)的zro6八面体的骨架构成。在石榴石型结构中,li原子可能存在于24d四面体位点或48g和96h八面体位点。这些原子中的大多数可能存在于这些位点。
[0067]
掺杂剂a本身可以占据li或zr位点。认为掺杂剂al、ga或fe通常在li位点。认为掺杂剂nb、w和ta通常在zr位点。
[0068]
无机化合物m优选具有立方结构。使用x射线衍射确定结构。该结构通常被描述为属于3-空间群。该结构也可以属于i-43d空间群,特别是当a=ga、fe或al ga时。
[0069]
无机化合物m是使用本领域技术人员已知的llzo石榴石制备技术制备的。可以参考在journal of the korean ceramic society[韩国陶瓷协会杂志]2019;56(2):111-129(doi:https://doi.org/10.4191/kcers.2019.56.2.01)中通过引用给出的方法。例如,可以使用固态方法制备它,通过该方法将氧化物或氧化物的组成元素的盐紧密混合,然后将获得的混合物在高温、典型地高于900℃下煅烧。更特别地,可以使用例如ep 2353203 b1中描述的方法,该方法包括以下步骤:(1)将li2co3、la(oh)3、zro2和至少一种元素a的氧化物、碳酸盐、氢氧化物或盐例如通过在液体介质(如乙醇)中研磨而紧密混合;(2)将获得的混合物在空气中在至少900℃的温度下煅烧至少1小时的时间段;(3)将li2co3与煅烧产物紧密混合,例如通过在液体介质(如乙醇)中研磨;(4)将获得的混合物在空气中在至少900℃的温度下煅烧,然后在至少1100℃的温度下煅烧。通常,将元素a的氧化物用于该合成。可以使用适用于任何具有式(i)的组合物的ep 2353203 b1的实例1的精确条件。也可以使用在j.mater.chem.a [材料化学杂志a],2014,2,172(doi:10.1039/c3ta13999a)中描述的固态方法,该方法包括以下步骤:(1)将li2co3、la(oh)3、zro2和至少一种元素a的氧化物、碳酸盐、氢氧化物或盐紧密混合;(2)将获得的混合物在空气中在至少1000℃的温度下煅烧至少10小时;(3)然后将煅烧的产物用研钵研磨并筛分以仅回收《75mm的颗粒,然后将这些颗粒在异丙醇中研磨。
[0070]
还可以使用共沉淀方法制备无机化合物m,通过该方法使包含元素la、zr和a的盐的溶液(例如conitrate的溶液)与碱性溶液接触,以获得沉淀物,然后使该沉淀物与锂盐接触并且在至少900℃的温度下煅烧沉淀物/锂盐混合物。可以使用适用于任何具有式(i)的组合物的us 2019/0051934的实例1的精确条件。
[0071]
其他方法描述于以下文献中:jp 2012-224520、us 2018/0248223、us 2019/0051934或ep 3135634 b1(具体参见实例1)。
[0072]
具有式(i)的无机化合物m包含具有式(ii)的氧化物或基本上由其组成:
[0073]
li
x1
la3zr
zawo12
ꢀꢀ
(ii)
[0074]
其中a、z和w如上所述,并且x1是正实数,其使得确保该氧化物的电中性。
[0075]
x、z和w如上所述。关于实数x1,其使得确保氧化物的电中性。为此,还考虑了除锂以外的氧化物的组成元素(即元素zr、la和a以及任选地hf)的比例。为了计算x1,还考虑了以下氧化态:li i;zr iv;hf iv;la iii;al iii;ga iii;nb v;fe iii;w vi;ta v。例如,对于由元素li、al、la和zr组成的氧化物,其中z=1.99且w=0.22(如通过化学分析给出),x1等于6.38(x1=24
–
3x3
–
4x1.99
–
3x0.22)。
[0076]
应注意,在无机化合物m的制备中,在高温下进行的一个或多个煅烧步骤具有使锂挥发的作用。为了补偿这一点,通常提供相对于具有式(i)的氧化物的化学计量过量的锂,使得x》x1。
[0077]
上文关于元素铪可能存在的描述也适用于具有式(ii)的氧化物。因此,应记住本发明因此也适用于具有式(iia)的氧化物:
[0078]
li
x1
la3(zr
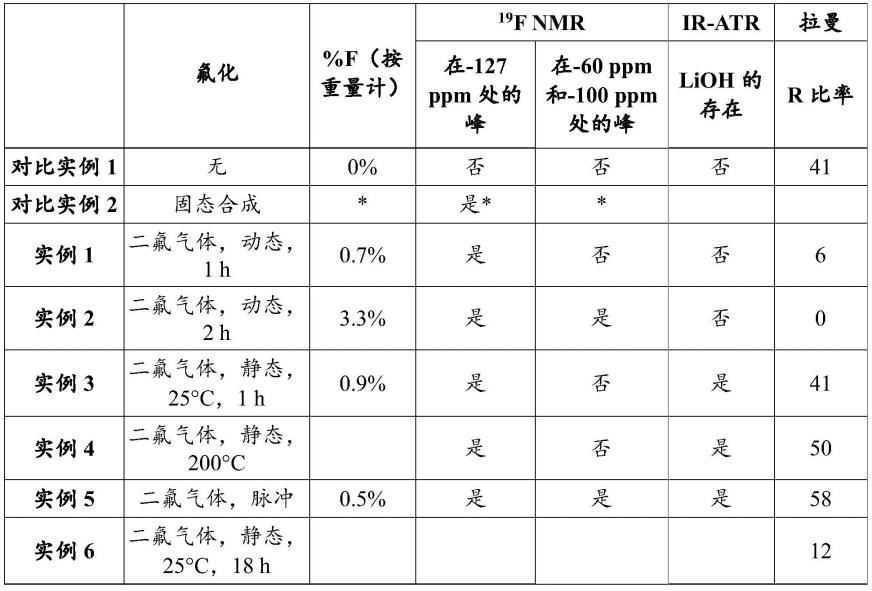
(1-a)
hfa)zo
12
ꢀꢀ
(iia)
[0079]
x1、z和a如上所述。
[0080]
具有式(ii)或者具有式(iia)的氧化物是石榴石型的。认为其结晶结构通常由lao8十二面体(la的配位数为8)的骨架和zro6八面体(zr的配位数为6)的骨架组成。更特别地,该结晶结构可以由配位数为8(24c位点)的lao8十二面体的骨架和配位数为6(16a位点)的zro6八面体的骨架构成。在石榴石型结构中,li原子可能存在于24d四面体位点或48g和96h八面体位点。这些原子中的大多数可能存在于这些位点。
[0081]
该氧化物优选具有立方结构。使用x射线衍射确定结构。该结构通常被描述为属于3-空间群。该结构也可以属于i-43d空间群,特别是当a=ga、fe或al ga时。
[0082]
通过使无机化合物m(并且因此具有式(ii)的氧化物)与包含二氟(f2)气体的气氛接触进行氟化。
[0083]
氟化气氛可以基本上由二氟气体构成。二氟气体在气氛中的比例大于99.0%、或甚至99.5%、或甚至99.9%。所有这些比例均以体积%表示。实例中给出了包含二氟气体的气氛的实例。
[0084]
氟化对应于在固体与气体之间的反应。其可以以静态模式进行,根据该模式,将无机化合物m和氟化气氛引入优选预先置于真空下的密封腔室中,并使其反应。在预先置于真空下的情况下,可以施加至少10-2
毫巴的低真空。可以施加在100与500毫巴之间的初始f2压力。还可以参考文章“fluorinated nanodiamonds as unique neutron reflector[氟化纳米金刚石作为独特的中子反射器]”,carbon[碳],第130卷,2018年4月,第799-805页中描述的氟化程序,并且还参考实例。根据上述静态模式的变体(“脉冲”模式),将腔室中的氟化气氛分几次引入含有无机化合物m的密封腔室中,并且在两次添加之间,使氟化气氛与固体反应。静态模式及其变体可以根据实例中详细描述的方案进行(分别参见实例3-4和实例5)。
[0085]
氟化方法也可以有利地以动态模式进行,根据该模式,将氟化气氛连续地引入含有无机化合物m的开放腔室中。流入开放腔室的氟化气氛的体积流速(在20℃和大气压下测量)可以在10与100ml/min之间、更特别地在10与30ml/min之间。还可以参考文章“the synthesis of microporous carbon by the fluorination of titanium carbide[通过碳化钛的氟化合成微孔碳]”,carbon[碳],第49卷,第9期,2011年8月,第2998-3009页中描述的程序。动态模式可以根据实例1和2中详细描述的方案进行。
[0086]
无论使用何种模式,在氟化结束时,将过量的二氟气体(像反应的产物)用惰性气
体(例如像n2或he)吹扫并在位于反应器下游的碱石灰捕集器中中和。
[0087]
无论使用何种模式,固体与氟化气氛之间的接触的总持续时间在2分钟与4小时之间、或甚至在2分钟与2小时之间、或甚至在30分钟与2小时之间。
[0088]
氟化是在可变的温度下进行的。该温度可以在20℃与300℃之间、优选在20℃与250℃之间。其优选在“低”温度下、优选在20℃与50℃之间进行,以便不降低氧化物的物理化学特性、特别是导电性。
[0089]
当然,从实用的观点来看,无论何种模式,都优选使用耐二氟气体腐蚀的腔室。因此,腔室的材料必须是耐腐蚀的,这使得还可以防止由在其表面处存在的元素引起的任何污染。可以有利地使用由nif2钝化的镍制成的腔室。可以将固体放置在插入腔室中的同样由钝化镍制成的板上。
[0090]
为了促进固体与气体之间的接触,可以将固体以床的形式布置,其厚度可以有利地小于或等于5mm。无机化合物m优选呈粉末的形式,以促进与氟化气氛的接触。该粉末可以具有小于50μm、更特别地小于30μm的d50。d50对应于通过激光衍射技术对固体在液体介质中、特别是在水中的分散体获得的尺寸分布(按体积计)的中值直径。
[0091]
关于氟化无机化合物
[0092]
本发明还涉及在上述方法结束时获得的氟化无机化合物。该化合物的化学组成基本上对应于由上面给出的化学式之一给出的化学组成,应当理解,该化合物还包含元素氟。
[0093]
因此,本发明还涉及一种无机化合物,其具有石榴石型结构并且基于元素o、li、zr、a和任选地hf,这些元素的相对比例是具有式(i)的那些,该化合物还包含元素f并且具有以下特征中的至少一项:
[0094]
●
在(
19
f)固态nmr光谱上,信号位于-125.0与-129.0ppm之间、更特别地-126.0与-128.0ppm之间、还更特别地-126.5与-127.5ppm之间,在δ=0ppm处的参考是化合物cf3cooh的参考;
[0095]
●
比率r小于或等于50%、更特别地小于或等于40%、还更特别地小于或等于30%或20%或10%,r是在位于约1090cm-1
处的碳酸根基团的c-o键的振动带(对称拉伸ν)强度与位于约648cm-1
处的zro6八面体中的键的拉伸带强度之间的比率,这两个强度通过拉曼光谱确定。
[0096]
下面给出了关于该无机化合物的表征的进一步细节。
[0097]-通过(
19
f)固态nmr表征
[0098]
无机化合物的(
19
f)固态nmr光谱可以具有位于-125.0与-129.0ppm之间、更特别地-126.0与-128.0ppm之间、还更特别地-126.5与-127.5ppm之间的信号。化学位移通过在δ=0ppm处取cf3cooh作为参考给出。该信号总体上是对称的。该信号总体上归因于在li-f键中涉及的氟。
[0099]
nmr光谱可以有利地用30khz的魔角旋转获得。
[0100]
更特别地,可以使用实例中给出的测量条件。
[0101]
通过相同的光谱技术且在相同的条件下,还可以观察到在-98.0与-102.0ppm之间、更特别地在-99.0与-101.0ppm之间、还更特别地在-99.5与-100.5ppm之间的信号,和/或在-58.0与-62.0ppm之间、更特别地在-59.0与-61.0ppm之间、还更特别地在-59.5与-60.5ppm之间的信号。这些信号通常分别归因于la-f和zr-f键的形成。
[0102]-通过拉曼光谱表征
[0103]
氟化作用也可以使用拉曼光谱来证明。因此,氟化无机化合物具有小于或等于50%、更特别地小于或等于40%、还更特别地小于或等于30%或20%或10%的比率r,r是在位于约1090cm-1
处的碳酸根基团的c-o键的振动带(对称拉伸ν)强度与位于约648cm-1
处的zro6八面体中的键的拉伸带强度之间的比率。
[0104]
通常认为碳酸根基团的c-o振动带位于1090
±
20cm-1
处。此带总体上位于1080与1100cm-1
之间。
[0105]
通常认为zro6八面体的拉伸带位于648
±
20cm-1
处。此带总体上位于638与658cm-1
之间。
[0106]
此外,观察到在充气的密封烧瓶中储存至少两个月、特别是两个月的时间段后,无机化合物可以具有相同的r比率。
[0107]-通过红外光谱以衰减全反射(atr)模式进行表征
[0108]
氟化作用也可以使用红外光谱以衰减全反射(atr)模式来证明。具体地,碳酸根基团具有分别位于1350与1600cm-1
之间和890与1350cm-1
之间的振动模式ν3和ν2。因此,碳酸根基团的振动模式ν3和/或振动模式ν2的强度小于或等于50%、更特别地小于或等于40%、还更特别地小于或等于30%或20%或10%,这些模式分别位于1350与1600cm-1
之间和890与1350cm-1
之间。
[0109]
关于r比率,在充气的密封烧瓶中储存至少两个月、特别是两个月的时间段后,无机化合物可以具有该相同的强度。
[0110]-氟的比例
[0111]
氟在化合物中的比例(以元素氟的重量相对于总重量表示)通常小于或等于10.0%、更特别地小于或等于7.0%、还更特别地小于或等于5.0%。该比例通常大于或等于0.01%、更特别地大于或等于0.10%、还更特别地大于或等于0.50%。该比例可以在0.01%与10.0%之间、更特别地在0.10%与10.0%之间、或甚至在0.10%与7.0%之间。该比例可以使用百分制分析或者通过
19
f nmr确定。为了通过nmr确定氟的比例,可以使用含有元素氟的内标物,其信号与无机化合物的信号不一致。例如,可以使用pvdf均聚物。对于pvdf标准物,可以特别地使用下式:
[0112][0113]
其中a1为pvdf的氟信号的面积总和,m1为pvdf的质量,a2为无机化合物的氟信号的面积总和,m2为无机化合物的质量,并且[f]
pvdf
为pvdf中氟的按质量计的浓度,即59。
[0114]
观察到氟化方法具有减少特别是在固体表面处存在的碳酸根基团的量、或甚至使它们消失的作用。这种减少/消失是逐渐的,特别地取决于固体与氟化气氛之间的接触时间。因此,本发明的方法可以使固体表面脱碳,这确保了对其的有效保护,特别是甚至在将固体在露天储存之后。
[0115]
氟化是在“温和”条件下进行的,使得起始固体的结晶结构不会受到不利影响。换句话说,氟化无机化合物具有与起始固体相同的结晶结构。因此,其优选具有立方结构。使用x射线衍射确定结构。该结构通常被描述为属于3-空间群。该结构也可以属于i-43d空间群,特别是当a=ga、fe或al ga时。此外,氟化无机化合物通常由lao8十二面体(la的配位数
为8)的骨架和zro6八面体(zr的配位数为6)的骨架组成。更特别地,该结晶结构可以由配位数为8(24c位点)的lao8十二面体的骨架和配位数为6(16a位点)的zro6八面体的骨架构成。在石榴石型结构中,li原子可能存在于24d四面体位点或48g和96h八面体位点。
[0116]
此外,氟化通常不会导致x射线衍射峰变宽。
[0117]
氟化无机化合物的用途
[0118]
氟化无机化合物可以用作锂电池的固体电解质。它也可以用于制备锂电池。氟化无机化合物可以用于制备电极e。电极e可以是正极(e
p
)或负极(en)。
[0119]
电极e典型地包含:
[0120]
●
金属载体;
[0121]
●
与金属基底接触的组合物(c)的层,所述组合物(c)包含:
[0122]
(i)所述的氟化无机化合物;
[0123]
(ii)至少一种电活性化合物(eac);
[0124]
(iii)任选地,除氟化氧化物(licm)之外的至少一种传导li离子的材料;
[0125]
(iv)任选地至少一种导电材料(ecm);
[0126]
(v)任选地锂盐(lis);
[0127]
(vi)任选地至少一种聚合物粘合剂材料(p)。
[0128]
术语电活性化合物(eac)表示可以将锂离子掺入其结构中并在电池的充电和放电期间释放它们的化合物。eac的性质取决于它是正极还是负极而变化:
[0129]
1)正极e
p
[0130]
eac可以是具有式limeq2的硫属化物型化合物,其中:
[0131]-me表示选自由co、ni、fe、mn、cr、al和v形成的组的至少一种金属;
[0132]-q表示o或s。
[0133]
eac可以更特别地具有式limeo2。下面给出了eac的实例:licoo2、linio2、limno2、lini
x
co
1-x
o2(0《x《1)、lini
x
coymnzo2(0《x,y,z《1且x y z=1)、li(ni
x
coyalz)o2(x y z=1)以及具有尖晶石型结构的化合物limn2o4和li(ni
0.5
mn
1.5
)o4。
[0134]
eac还可以是具有式m1m2(jo4)
fe1-f
的锂化或部分锂化的化合物,其中:
[0135]-m1表示锂,其可以被另一碱金属部分取代;
[0136]-m2表示 2氧化态的过渡金属,其选自fe、mn、ni或这些元素的组合,其可以被至少一种氧化态在 1与 5之间的其他过渡金属部分取代;
[0137]-jo4表示氧阴离子,其中j选自由p、s、v、si、nb、mo或这些元素的组合组成的列表;
[0138]-e表示f、oh或cl;
[0139]-f表示jo4氧阴离子的摩尔分数,并且可以在0.75与1之间。
[0140]
eac还可以是硫或li2s。
[0141]
2)负极en[0142]
eac可以选自由能够在其结构中容纳锂的石墨碳形成的组。该类型的eac的进一步细节可以在carbon[碳]2000,38,1031-1041中找到。该类型的eac通常以粉末、薄片、纤维或球体的形式存在。
[0143]
eac还可以是锂金属;基于锂的化合物(例如像us 6,203,944或wo00/03444中描述的那些);钛酸锂,通常由式li4ti5o
12
表示。
[0144]
ecm典型地选自导电碳基化合物的组。这些碳基化合物例如选自由炭黑、碳纳米管、石墨、石墨烯和石墨纤维形成的组。例如,它们可以是炭黑,如科琴黑或乙炔黑。
[0145]
lis可以选自由以下形成的组:lipf6、双(三氟甲磺酰基)亚胺锂、双(氟磺酰基)亚胺锂、lib(c2o4)2、liasf6、liclo4、libf4、lialo4、lino3、licf3so3、lin(so2cf3)2、lin(so2c2f5)2、lic(so2cf3)3、lin(so3cf3)2、lic4f9so3、licf3so3、lialcl4、lisbf6、lif、libr、licl、lioh和2-三氟甲基-4,5-二氰基咪唑锂。
[0146]
聚合物粘合剂材料(p)的功能是将组合物(c)的组分粘合在一起。它通常是惰性材料。它优选是化学稳定的并且必须允许离子传输。下面给出了材料p的实例:基于偏二氟乙烯(vdf)的聚合物和共聚物、苯乙烯-丁二烯弹性体(sbr)、sebs型的共聚物、聚(四氟乙烯)(ptfe)和pan型的共聚物。优选地,它是基于vdf的聚合物或共聚物,例如pvdf,或者基于vdf和至少一种除vdf以外的氟化共聚单体(如六氟丙烯(hfp))的共聚物。
[0147]
组合物(c)中氟化无机化合物的比例可以在按重量计0.1%与80%之间,该比例以氟化氧化物的重量相对于组合物的总重量表示。此比例可以是按重量计在1.0%与60.0%之间、或者甚至按重量计在10.0%与50.0%之间。
[0148]
电极(e)的厚度没有限制,并且应当适配于预期应用所需要的能量和功率。因此,该厚度可以在0.01与1000mm之间。
[0149]
氟化无机化合物还可以用于制备电池隔膜(sp)。隔膜表示在电池的正极与负极之间的渗透膜。它的功能是可透过锂离子,同时阻挡电子,并且同时确保电极之间的物理分隔。本发明的隔膜(sp)典型地包含:
[0150]
●
氟化无机化合物;
[0151]
●
至少一种聚合物粘合剂材料(p);
[0152]
●
至少一种金属盐,例如锂盐;
[0153]
●
任选地增塑剂。
[0154]
电极(e)和隔膜(sp)可以使用本领域技术人员已知的技术制备。这些技术通常包括在适当的溶剂中混合各组分,并且然后除去该溶剂。因此,例如,电极(e)可以通过包括以下步骤的方法来制备:
[0155]-将包含组合物(c)的各组分和至少一种溶剂的分散体施加在金属载体上;
[0156]-然后除去溶剂。
[0157]
可以使用杂志energy environ.sci.[能量环境科学],2019,12,1818中描述的用于制备电极(e)和隔膜(sp)的技术。
[0158]
如果通过援引并入本文的任何专利、专利申请和出版物的披露内容与本技术的说明相冲突到了可能导致术语不清楚的程度,则本说明应优先。
[0159]
实例
[0160]
19
f nmr光谱
[0161]
在具有魔角旋转(mas)的bruker 400mhz固态avance neo或bruker 300mhz avance光谱仪上以30或26khz的旋转速率进行
19
f核的固态nmr。化学位移相对于cf3cooh(δ=0ppm)作为参考(观察:δ(cf3cooh)=-78.5ppm相对于cfcl3)。
[0162]
测量条件:使用单个π/2脉冲,循环延迟d1为30s。调整脉冲的数量以获得高信噪比(典型地128或256个脉冲)。
[0163]
氟测定
[0164]
通过
19
f mas-nmr(魔角旋转)进行氟的定量。使用配备有1.9mm探针的bruker 400mhz avance neo光谱仪。将pvdf(来自索尔维公司(solvay)的1015/1001)用作内标物并且所使用的
19
f参考物是三氟乙酸(δ=0ppm)。
[0165]
使用精密天平称量从25至80mg的样品和从2至5mg的pvdf。使用粉末混合器,例如通过在来自wab的混合器中三维混合5分钟,制备这两种固体的均匀混合物。将约25mg的该混合物在1.9mm转子中压实并引入光谱仪中。用自旋速率为26khz、脉冲为1.5ms且弛豫时间d1为30秒的单脉冲序列分析样品。
[0166]
通过积分nmr notebook上的信号来分解nmr光谱。pvdf信号(主信号和旋转带)的面积相加,其方式与归因于样品中存在的氟的信号相同。
[0167]
氟在样品中的重量百分比根据下式给出:
[0168][0169]
其中a1为pvdf的氟信号的面积总和,m1为pvdf的质量,a2为无机化合物的氟信号的面积总和,m2为无机化合物的质量,并且[f]
pvdf
为pvdf中氟的按质量计的浓度,即59。
[0170]
拉曼光谱
[0171]
在来自jobin-yvon公司的配备有共聚焦显微镜的t64000光谱仪上通过拉曼光谱分析产物。在环境条件下在密封烧瓶中储存2个月后记录光谱。所使用的入射激光是在514.5nm下操作的离子化氩激光。激光的入射功率为100mw。在250-1500cm-1
范围内进行拉曼分析,对于光谱宽度为500cm-1
的每个窗口,采集时间为60s,重复两次。
[0172]
以atr模式的红外光谱
[0173]
使用nicolet 380ft-ir(热-电子)傅立叶变换分光光度计记录在400与4000cm-1
之间的ir光谱。在环境条件下在密封烧瓶中储存2个月后记录光谱。每个光谱由128次扫描组成,分辨率为4cm-1
。设备会自动减去背景。
[0174]
扫描电子显微镜-能量色散光谱分析(sem-eds)
[0175]
用于sem-eds表征的操作程序:
[0176]
将粉末包埋在epofix树脂中,该树脂在室温下聚合超过24h。
[0177]
聚合后,将含有粉末的固体块在干燥条件下在切片机装置(reichert&jung ultracut e型)上切片;因此,一些固体颗粒的截面是可以得到的。
[0178]
然后在cressington 208hr溅射涂布机中,在二次真空下,通过铂溅射处理制备物的表面。沉积厚度为几纳米。
[0179]
将制备物引入sem feg leo 1525中。在8kv下进行sem eds分析,其中隔板为60μm且工作距离为8.5mm。通过由珀耳帖效应冷却的oxford sdd 80mm2检测器x max n分析eds光谱。在硅标准物上进行光束优化后,在aztec软件v4.4下进行数据处理。在放大2000倍下获得线轮廓,在典型地为20μm的长度上有500个数据点。在8kv的条件下,分析体积≤1μm3。
[0180]
元素f(k线)和la(m线)的绝对强度被测量为线轮廓上的位置的函数。结果报告在图3和图4中。
[0181]
x射线光电子能谱(xps)
[0182]
通过xps进行用于表面元素分析的操作程序:
[0183]
将粉末样品压在铟球上。xps仪器是具有单色化alkαx射线源的thermo k-α 。数据处理软件是avantage。由每种元素的高分辨率光谱获得原子浓度。
[0184]
在样品“原样”上进行xps分析,并且还在通过ar
离子蚀刻之后进行。为了给出溅射厚度的数量级,我们在下面的结果中参考sio2的溅射速率。
[0185]
分析和仪器规格如下:
[0186]-出射角:90
°
;
[0187]-分析深度:小于10nm(平均3nm);
[0188]-光斑直径:400μm;
[0189]-除h和he之外,所有元素均可检测;
[0190]-灵敏度极限:按原子计从0.1%至0.5%;
[0191]-定量分析:
[0192]
*准确度10%至20%;
[0193]
*精确度为2%至5%(相对),取决于浓度。
[0194]
xps分析的结果报告于表2中。
[0195]
使用的无机化合物m是通过在j.mater.chem.a[材料化学杂志a]2014,2(1),172
–
181.(https://doi.org/10.1039/c3ta13999a)中描述的方法获得的al掺杂的llzo。阳离子具有通过icp确定的以下相对组成:li
6.97
la3zr
1.98
al
0.22
。
[0196]
对于氟化,使用99.9%纯的二氟气体(f2)的气氛(hf杂质水平《0.5vol%并且o2/n2杂质水平为约0.5vol%)。
[0197]
实例1:在环境温度下以动态模式氟化1小时
[0198]
将336.5mg m以粉末床的形式沉积在钝化的镍舟中,其厚度小于2mm。将板在25℃下插入1-升钝化的镍反应器中。在1小时内将20ml/min的f2流连续引入反应器中。在试验结束时,在60分钟内使用50ml/min的n2流吹扫反应器中任何痕量的残留f2。实验后观察到1.1mg的质量吸收,表示氟掺入化合物m中。
[0199]
实例2:在环境温度下以动态模式氟化2小时
[0200]
重复实例1的条件,m的初始质量为402.7mg m,并且时间为2小时而不是1h。实验后观察到1.8mg的质量吸收,表示氟掺入化合物m中。
[0201]
实例3:在环境温度下以静态模式氟化1小时
[0202]
将约500mg化合物m作为厚度小于2mm的粉末床沉积在钝化的镍舟中。将板在25℃下插入1升反应器中。在1小时内在反应器中施加200毫巴的f2压力。反应器中的温度不受监测,并且对应于环境温度,约为25℃。在试验结束时,在60分钟内使用50ml/min的n2流吹扫反应器中任何痕量的f2。
[0203]
实例4:在200℃下静态氟化
[0204]
将509.3mg化合物m作为厚度小于2mm的粉末床沉积在钝化的镍舟中。将板在25℃下插入1升反应器中。在整个实验期间在反应器中施加200毫巴的f2压力。监测反应器的温度,并且以2℃/min的速率升至200℃,然后使反应器在n2流(50ml/min)下自由冷却至环境温度,即约1h 30min。
[0205]
实例5:以脉冲模式氟化
[0206]
将592.2mg化合物m作为厚度小于2mm的粉末床沉积在钝化的镍舟中。将板在25℃下插入1升反应器中。在反应器中每2分钟计量添加20毫巴f2,直到达到200毫巴的压力。在试验结束时,使用50ml/min的n2流持续60分钟吹扫反应器中任何痕量的f2。实验后观察到5.0mg的质量吸收,表示氟掺入化合物m中。
[0207]
实例6:在环境温度下以静态模式氟化18小时
[0208]
将约500mg化合物m作为厚度小于2mm的粉末床沉积在钝化的镍舟中。将板在25℃下插入1升反应器中。在3个步骤中在反应器中施加1000毫巴的f2压力。反应器中的温度不受监测,并且对应于环境温度,约为25℃。
[0209]
1)以500ml/min的流速向反应器中添加500毫巴的n2;
[0210]
2)以150ml/min的流速向反应器中添加200毫巴的f2;
[0211]
3)以500ml/min的流速向反应器中添加300毫巴的n2。
[0212]
进行这3个步骤需要14分钟。在整个实验过程中,即在18h期间,在反应器中施加1000毫巴的反应混合物(按体积计20%f2/80%n2)压力。
[0213]
在试验结束时,在60分钟内使用500ml/min的n2流吹扫反应器中任何痕量的f2。
[0214]
对比实例1:化合物m不经任何氟化而使用。
[0215]
对比实例2:通过固态合成制备氟化llzo
[0216]
通过混合5.24g的li2co3(99,9%,西格玛奥德里奇公司(sigma aldrich))、9.72g的la2o3(99%,西格玛奥德里奇公司)、4.93g的zro2(西格玛奥德里奇公司)、0.21g的al2o3(西格玛奥德里奇公司,在600℃下预煅烧2h)、和0.51g的lif(西格玛奥德里奇公司)进行固态合成。目标化学计量为li
6.4
la3al
0.2
zr2o
12
1.5lif,并且因此目标氟含量为3.3%wt。
[0217]
步骤1:将粉末与66g的5mm氧化锆球(预先在65℃的烘箱中干燥)混合,并且置于turbula装置中2小时以使它们均匀。
[0218]
然后将球与粉末分离,并且将粉末置于覆盖有氧化铝盖的氧化铝坩埚(矩形)中。
[0219]
然后在12小时期间在空气下将粉末在900℃下煅烧,升温速率为5℃/min并且冷却速率为2℃/min,然后在100℃保持平稳,以避免在煅烧结束时的任何水分吸收,然后在50℃下回收。
[0220]
步骤2:将所得粉末在turbula中与66g的5mm氧化锆球(在65℃的烘箱中干燥)混合。
[0221]
然后将球与粉末分离,并且将粉末置于覆盖有氧化铝盖的氧化铝坩埚(矩形)中。
[0222]
然后在12小时期间将粉末在1000℃下煅烧,升温速率为5℃/min并且冷却速率为2℃/min,然后在100℃保持平稳,以避免在煅烧结束时的任何水分吸收,然后在50℃下回收
[0223]
步骤3:将所得粉末在turbula中与66g的5mm氧化锆球(在65℃的烘箱中干燥)混合。
[0224]
然后将球与粉末分离,并且将粉末置于覆盖有氧化铝盖的氧化铝坩埚(矩形)中。
[0225]
然后在12小时期间在炉f1300中将粉末在1100℃下煅烧,升温速率为5℃/min并且冷却速率为2℃/min,然后在100℃保持平稳,以避免在煅烧结束时的任何水分吸收,然后在50℃下回收。
[0226]
样品的xrd表明存在立方llzo(95%wt,通过highscore软件测量)和少量痕量的la2zr2o7(5%wt,通过highscore软件测量)。
[0227]
表i中给出了一些结果。可以得到以下观察结果:
[0228]
19
f nmr:在与氟化气氛接触的所有样品上,在-127
±
2ppm处观察到相对于cf3cooh的对称信号,这证明了li-f键的存在。对于某些样品,相对于cf3cooh,在-100ppm和-60ppm处出现另外的信号,表示出现了
19
f核的新化学环境。
[0229]
拉曼光谱:所有光谱都具有立方llzo的特征峰,即:
[0230]-在354cm-1
和508cm-1
处的峰是基于zro6的八面体单元的变形模式的特征;
[0231]-在648cm-1
处的峰是这些相同单元的拉伸模式的特征。
[0232]
在除实例2的产物之外的所有产物上在约1090cm-1
处也可以观察到另外的峰。该峰归因于碳酸根基团的c-o键的(对称拉伸)振动(参见zhang z,zhang l,liu y,wang h,yu c,zeng h,wang lm,xu b,interface-engineered li7la3zr2o
12-based garnet solid electrolytes with suppressed li-dendrite formation and enhanced electrochemical performance[具有抑制的li-枝晶形成和增强的电化学性能的界面工程化的基于li7la3zr2o
12
的石榴石固体电解质],chemsuschem[化学与可持续性、能源与材料]2018,11,3774-3782)。
[0233]
ir-atr光谱:主要振动模式与碳酸根在1409-1460cm-1
和879cm-1
处的振动ν3和ν2有关。对于以动态模式处理2h的llzo,这些带几乎完全消失。有时在某些产物中观察到在626、679、847、1002、2800和3613cm-1
处的一系列带,并且表明lioh的存在。
[0234]
表i
[0235][0236]
*通过
19
f固态nmr,证明了以约-40ppm为中心的另外的峰。它是根据本发明的实例的产物中不存在的宽信号,这归因于在通过固态合成获得的产物中的不同
19
f环境。该信号的存在阻止了通过上述nmr定量方法对氟含量进行定量。在约-127ppm处存在峰。
[0237]
观察到与静态模式相比,动态模式使得可以获得具有非常低的r比率的固体。然而,当增加静态模式下的反应时间时,也可以获得具有低r比率的固体(参见实例6)。
[0238]
在动态模式下,观察到所得固体中f的重量%随着反应时间而增加(参见实例1和实例2),并且因此,结论是至少通过监测反应时间,可以控制样品中的氟含量。
[0239]
如图3和图4所报告,sem-eds分析的结果表明氟和镧元素沿氟化llzo固体颗粒的截面分布,这些固体颗粒分别通过根据实例2的气态氟化和根据对比实例2的固相合成制备。一方面,图3显示,在通过气态氟化制备的氟化llzo固体颗粒的情况下,氟集中在颗粒的表面上,而颗粒的核是“富镧的”。另一方面,图4显示,当通过固相合成制备氟化llzo固体颗粒时,氟和镧全都沿颗粒的截面均匀分散。
[0240]
诸位申请人已经表明,气态氟化允许有利地进行主要位于llzo颗粒表面上的氟化。
[0241]
xps分析的结果报告在下表2中。比率c/zr和f/zr以材料深度的函数表示。结果表明,碳/c含量(c被假定来自通过ir和/或拉曼光谱鉴定的碳酸盐物质)随着材料深度而降低。更出人意料地,无论分析深度如何,相对于对比实例1,实例2的氟化产物的c含量低得多。这与实例2的产物中较高的氟量直接相关。
[0242]
该结果与以下结论一致,即根据本发明的氟化方法具有减少特别是在固体表面处存在的碳酸根基团的量的作用。
[0243]
值得注意的是,实例2中的氟含量随深度而降低,这与sem-eds结果非常一致。
[0244]
表2
[0245][0246]
上述证据表明:i)气态氟化允许有利地进行主要位于llzo颗粒表面上的氟化,ii)该氟化伴随着所述表面上的碳酸根物质的去除,以及iii)该氟化保护所述表面不进一步形成碳酸根。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。