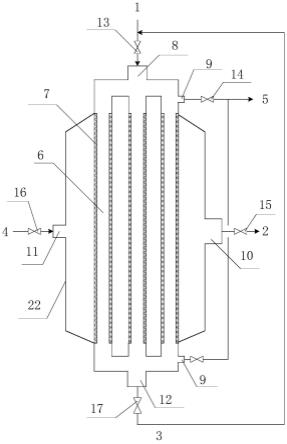
1.本发明属于丙烯酸合成方法的技术领域,具体涉及一种丙烯酸合成用催化剂及其制备方法和丙烯酸合成用成型催化剂及其制备方法和丙烯酸合成方法。
背景技术:
2.丙烯酸及其酯广泛应用于建筑、电子、汽车工业等领域,合成涂料、胶粘剂、吸水性树脂等,2020年全球丙烯酸产能超过850万吨/年,中国丙烯酸产能达318万吨/年,每年国内丙烯酸催化剂需求量约1000吨。
3.目前工业上制丙烯酸使用丙烯两步氧化法,在第一反应器中,mo-bi复合氧化物催化剂作用下,丙烯氧化生成丙烯醛,在第二反应器中,mo-v复合氧化物催化剂作用下,丙烯醛氧化生成丙烯酸,同时生成co、co2、乙醛和乙酸等副产物,并放出大量热量。
4.目前常用的制备上述催化剂的方法多采用将金属化合物制成混合液,再加入不溶性氧化物进行蒸发,然后煅烧、粉碎和成型的过程,通过这些制备方法获得的催化剂通常缺少稳定的晶体形态和特殊的孔径分布,使用寿命较短,催化剂的转化率和选择性较低,产品的收率较低。
技术实现要素:
5.本发明所要解决的技术问题之一是针对现有技术中丙烯醛氧化合成丙烯酸的反应中所用催化剂孔径分布不合理,产物收率低的缺陷,提供一种具有晶体形态均一、稳定性高、使用寿命长的多级孔分布的、丙烯酸收率高的催化剂。
6.根据本发明的第一方面,本发明提供一种丙烯酸合成用催化剂,所述催化剂具有通式mo
12va
cubog的活性组分,其中,
7.a=0.5~10;
8.b=0.1~8;
9.g为由所述通式中除氧之外的元素的总化合价决定的数值;
10.所述催化剂具有mo3vo
11
晶相结构,且mo3vo
11
具有定向生长的特征,催化剂的xrd谱图中,特征峰2θ=24.9(540晶面)与2θ=22.2(001晶面)的峰强度比范围为0-0.65,优选为0.1-0.4。
11.优选地所述催化剂具有通式mo
12va
cubwcmdog的活性组分,其中,
12.a=0.5~10;
13.b=0.1~6;
14.c=0.01~6;
15.d=0~5;
16.所述m选自ni、mn、sb、ca、fe、te和nb中的一种或多种;
17.和/或
18.催化剂的xrd谱图包括但不限于:2θ=31.5
°
(810晶面),2θ=28.2
°
(640晶面),2θ=24.9
°
(540晶面),2θ=23.3
°
(600晶面),2θ=22.2
°
(001晶面)。
19.根据本发明的第二方面,本发明提供一种丙烯酸合成用成型催化剂,所述成型催化剂含有活性组分和载体,所述活性组分为本发明所述催化剂。
20.优选地,以100质量份所述成型催化剂为基准,所述成型催化剂包括20~90质量份的活性组分和10~80质量份的载体。
21.优选地,所述载体选自sio2、al2o3、zro2和tio2中的一种或多种。
22.优选地,所述成型催化剂具有多级孔分布,包括:第一级孔径范围为2-50nm(指的是大于等于2nm至小于等于50nm)的小孔,其孔容占总孔容的比例范围为30-70%,第二级孔径范围为50-1000nm(指的是大于50nm至小于等于1000nm)的大孔,其孔容占总孔容的比例范围为30-70%,所述成型催化剂的总孔容范围为:0.001-0.1cm3/g,优选0.01-0.06cm3/g。
23.根据本发明的第三方面,本发明提供一种丙烯酸合成用催化剂的制备方法,该方法包括:
24.(1)将活性组分原料化合物溶于水中得到混合液a,将醇、水和长链有机酸混合均匀得到混合液b;
25.(2)将混合液a与混合液b混合并调节混合液中性至碱性,得到悬浊液c;
26.(3)将得到的悬浊液进行热处理,可选地进行粉碎。
27.优选地,所述醇为c1-c6的一元醇或多元醇,优选为甲醇、乙醇、1-丙醇、2-丙醇、1,3-丙二醇和丙三醇中的一种或多种,更优选为乙醇、1-丙醇和2-丙醇中的一种或多种,进一步优选为乙醇和1-丙醇的混合物,二者用量质量比为0.1-10:1。
28.优选地,所述长链有机酸为c8-c18的有机酸,优选为油酸、亚油酸、硬脂酸、软脂酸、辛酸、异辛酸、任酸、正癸酸、十一烯酸、月桂酸和十四酸中的一种或多种,更优选为油酸、亚油酸、硬脂酸和软脂酸中的一种或多种,优选为油酸和硬脂酸的混合物,二者用量质量比为1.5-5:1。
29.优选地,所述原料化合物包括活性组分元素的含氧盐和/或含氧盐水合物。
30.优选所述原料化合物包括七钼酸铵和/或其水合物、偏钒酸铵和/或其水合物、及硝酸铜和/或其水合物和偏钨酸铵和/或其水合物。
31.优选地,混合液b中,
32.水与乙醇的质量比为1:9至9:1,和/或油酸与水和醇总重的质量比为2:8至8:2;和/或
33.所述悬浊液c的ph值范围为7-12;和/或
34.步骤(2)中所述碱性调节剂为氨水、naoh、koh中的一种或多种,优选为氨水;和/或
35.步骤(3)的热处理条件包括:干燥温度为80-120℃。
36.优选地,该方法包括:
37.(1)将活性组分原料化合物溶于水中得到混合液a,将醇、水和长链有机酸混合均匀得到混合液b;
38.(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加氨水,调ph值,继续搅拌,得到悬浊液。
39.(3)将得到的悬浊液转移至烘箱中烘干后进行粉碎,得到催化剂。
40.根据本发明的第四方面,本发明提供一种制备本发明所述的成型催化剂的方法,该方法包括:
41.i)按照本发明所述的方法制备催化剂活性组分;
42.ii)将所述活性组分、可选地造孔剂、可选地润滑剂与载体原料混合后进行成型、焙烧;
43.优选地,
44.所述焙烧的温度为200~1000℃,和/或所述焙烧的时间为0.5~100小时;和/或
45.活性组分:造孔剂=30:1-2:1优选为20:1-4:1;
46.更优选地,
47.所述焙烧温度为400~700℃;和/或所述焙烧时间为3~10小时;
48.优选地,所述载体原料为sio2、al2o3、zro2、tio2、sio2前驱体、al2o3前驱体、zro2前驱体和tio2前驱体中的一种或多种。
49.根据本发明的第五方面,本发明提供一种丙烯酸合成方法,该方法包括:将丙烯醛与催化剂接触,所述催化剂为本发明所述的催化剂,或本发明所述的成型催化剂,优选地,所述接触的条件包括:
50.温度为240~320℃,和/或体积空速为100~150ml
·
h-1
·
g-1
;
51.优选地,所述反应温度为260-280℃,和/或所述体积空速为100-120ml
·
h-1
·
g-1
。
52.本发明的催化剂晶体形态均一、稳定性高、使用寿命长且具有多级孔分布,具有转化率高、丙烯酸收率高,寿命长等效果。
53.使用本发明的催化剂,能够降低催化反应所需温度大概20℃左右,这于工业具有很高的应用价值。
具体实施方式
54.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
55.本发明提供一种丙烯酸合成用催化剂,所述催化剂具有通式mo
12va
cubog的活性组分,其中,
56.a=0.5~10;
57.b=0.1~8;
58.g为由所述通式中除氧之外的元素的总化合价决定的数值;
59.所述催化剂具有mo3vo
11
晶相结构,且mo3vo
11
具有定向生长的特征,催化剂的xrd谱图中,特征峰2θ=24.9(540晶面)与2θ=22.2(001晶面)的峰强度比范围为0-0.65,优选为0.1-0.4。本发明的催化剂具有具有mo3vo
11
晶相结构,且mo3vo
11
具有定向生长的特征,催化剂的xrd谱图中,特征峰2θ=24.9(540晶面)与2θ=22.2(001晶面)的峰强度比范围为0-0.65,从而使得本发明的催化剂具有高转化率和高选择性的优势。
60.根据本发明,优选所述催化剂具有通式mo
12va
cubwcmdog的活性组分,其中,a=0.5~10;
61.b=0.1~6;
62.c=0.01~6;
63.d=0~5;
64.所述m选自ni、mn、sb、ca、fe、te和nb中的一种或多种。
65.采用本发明前述的催化剂,具有高转化率和高选择性优势。
66.根据本发明,催化剂的xrd谱图包括但不限于:2θ=31.5
°
(810晶面),2θ=28.2
°
(640晶面),2θ=24.9
°
(540晶面),2θ=23.3
°
(600晶面),2θ=22.2
°
(001晶面)。上述特征能够说明,本发明的催化剂具有mo3vo
11
晶相结构,且同时具有001晶面定向生长的优势。
67.具有前述物化性质的催化剂均可以实现本发明的目的,将其进行成型同样能够适合于作为丙烯酸合成用催化剂,针对本发明,特别提供一种丙烯酸合成用成型催化剂,所述成型催化剂含有活性组分和载体,所述活性组分为本发明所述催化剂。
68.根据本发明的一种优选实施方式,以100质量份所述成型催化剂为基准,所述成型催化剂包括20~90质量份的活性组分和10~80质量份的载体。
69.本发明对所述载体无特殊要求,催化剂领域常用载体均可以用于本发明,本发明以下例举但不局限于的载体选自sio2、al2o3、zro2和tio2中的一种或多种。
70.根据本发明的一种优选实施方式,所述成型催化剂具有多级孔分布,包括:第一级孔径范围为2-50nm的小孔,其孔容占总孔容的比例范围为30-70%;第二级孔径范围为50-1000nm的大孔,其孔容占总孔容的比例范围为30-70%;通过前述特殊孔结构能够实现本发明的催化剂用于丙烯酸合成时具有目标产物收率高等优势。
71.根据本发明的一种优选实施方式,所述成型催化剂的总比表面范围为:1-50m2/g。
72.根据本发明的一种优选实施方式,所述成型催化剂的总孔容范围为:0.001-0.1cm3/g,优选0.01-0.06cm3/g。
73.具有前述物化性质的催化剂均可以实现本发明的目的,对其制备方法无特殊要求,针对本发明,特别提供一种制备本发明所述的丙烯酸合成用催化剂的制备方法,该方法包括:
74.(1)将活性组分原料化合物溶于水中得到混合液a,将醇、水和长链有机酸混合均匀得到混合液b;
75.(2)将混合液a与混合液b混合并调节混合液至中性或碱性,得到悬浊液c;
76.(3)将得到的悬浊液进行热处理,可选地进行粉碎。
77.根据本发明的一种优选实施方式,所述醇为c1-c6的一元醇或多元醇,优选为甲醇、乙醇、1-丙醇,2-丙醇,1,3-丙二醇和丙三醇中的一种或多种,更优选为乙醇、1-丙醇和2-丙醇中的一种或多种,进一步优选为乙醇和1-丙醇的混合物,二者用量质量比为0.1-10:1;采用前述醇均可以实现本发明的目的,特别是当使用乙醇等时,能够使得本发明的制备方法制备的催化剂具有高转化率和选择性优势。
78.根据本发明的一种优选实施方式,所述长链有机酸为c8-c18的有机酸,更优选为油酸、亚油酸、硬脂酸、软脂酸、辛酸、异辛酸、任酸、正癸酸、十一烯酸、月桂酸和十四酸中的一种或多种,更优选为油酸、亚油酸、硬脂酸和软脂酸中的一种或多种,优选为油酸和硬脂酸的混合物,二者用量质量比为1.5-5:1;采用前述有机酸均可以实现本发明的目的,特别是当使用油酸等时,能够使得本发明的制备方法制备的催化剂具有高转化率和选择性优
势。
79.根据本发明的方法,对所述活性组分的原料化合物无特殊要求,常用的原料化合物均可以用于本发明,针对本发明,优选所述原料化合物包括活性组分元素的含氧盐和/或含氧盐水合物。
80.根据本发明的一种优选实施方式,优选所述原料化合物包括七钼酸铵和/或其水合物、偏钒酸铵和/或其水合物、及硝酸铜和/或其水合物和偏钨酸铵和/或其水合物。
81.根据本发明的一种优选实施方式,优选混合液b中,水与乙醇的质量比为1:9至9:1,和/或长链有机酸与水和醇总重的质量比为2:8至8:2,更优选水与乙醇的质量比为1:5至5:1,和/或长链有机酸与水和醇总重的质量比为2:6至6:2。
82.根据本发明的一种优选实施方式,所述悬浊液c的ph值范围为7-12,更优选为7-10。
83.根据本发明的一种优选实施方式,步骤(2)中所述碱性调节剂为氨水、naoh和koh中的一种或多种,优选为氨水。
84.根据本发明的一种优选实施方式,步骤(3)的热处理条件包括:干燥温度为80-120℃。
85.根据本发明的一种优选实施方式,本发明的方法包括:
86.(1)将活性组分原料化合物溶于水中得到混合液a,将醇、水和长链有机酸混合均匀得到混合液b;
87.(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加氨水,调ph值至中性或碱性,继续搅拌,得到悬浊液。
88.(3)将得到的悬浊液转移至烘箱中烘干后进行粉碎,得到催化剂。
89.本发明提供本发明所述的成型催化剂的制备方法,该方法包括:
90.i)按照本发明所述的方法制备催化剂活性组分;
91.ii)将所述活性组分、可选地造孔剂、可选地润滑剂与载体原料混合后进行成型、焙烧。
92.根据本发明,所述造孔剂、润滑剂的种类可选范围较宽,常用的种类均可以用于本发明,针对本发明,优选造孔剂例如羟丙基纤维素和/或石墨,润滑剂例如为石墨。本发明在此不进行详细描述。
93.根据本发明的一种优选实施方式,优选地,所述焙烧的温度为200~1000℃,和/或所述焙烧的时间为0.5~100小时。
94.根据本发明的一种优选实施方式,优选地,所述焙烧温度为400~700℃,更优选为450℃、500℃、550℃、600℃、650℃;和/或所述焙烧时间为1~30小时,所述焙烧时间为1小时、1.5小时、2小时、2.5小时、3小时、3.5小时、4小时、4.5小时、5小时、5.5小时、6小时、6.5小时、7小时、7.5小时、8小时、8.5小时、9小时、10小时、20小时、30小时。
95.根据本发明的一种优选实施方式,优选地,所述载体原料为sio2、al2o3、zro2、tio2、sio2前驱体、al2o3前驱体、zro2前驱体和tio2前驱体中的一种或多种。
96.本发明提供一种丙烯酸合成方法,该方法包括:将丙烯醛与催化剂接触,所述催化剂为本发明所述的催化剂,或本发明所述的成型催化剂。
97.根据本发明的一种优选实施方式,优选地,所述接触的条件包括:温度为240~320
℃,和/或体积空速为100~150ml
·
h-1
·
g-1
。
98.根据本发明的一种优选实施方式,优选地,所述反应温度为260-280℃,和/或所述体积空速为100-120ml
·
h-1
·
g-1
。
99.本发明中,成型的方法和条件无特殊要求,例如可以采用压片成型的方法,具体颗粒可以依据需要进行选择。
100.根据本发明的一种优选实施方式,所述钼原料化合物可以在其他活性组分化合物加入之后形成分散液后再加入。
101.本发明的催化剂晶体形态均一、稳定性高、使用寿命长且具有多级孔分布。具有转化率高、丙烯酸选择性高的效果。
102.使用本发明的催化剂,能够降低催化反应所需温度大概20℃左右,这于工业具有很高的应用价值。
103.下面结合实施例对本发明进行详细说明。应当理解,此处所描述的实施方式及实施例仅用于说明和解释本发明,其并不用于限定本发明。
104.本发明中,通过氮气吸脱附测试所得催化剂的孔结构。
105.本发明中,通过x射线衍射测试xrd图谱。
106.以下实施例中使用的催化剂评价方法为:
107.将反应物丙烯醛通入装填有待测催化剂的固定床反应器中,将反应后的产物用0℃稀酸吸收,再使用气相色谱进行分析,分析过程中计算碳平衡,选择碳平衡在95~105%时的数据为有效数据,其中,反应条件为:
108.反应器:固定床反应器,内径25.4毫米,长度750毫米;
109.催化剂填装量:150克;
110.反应温度:270℃;
111.反应时间:500小时;
112.原料体积比:丙烯醛:空气:水蒸气=1:3.2:2.1;
113.丙烯醛体积空速:110ml
·
h-1
·
g-1
。
114.实施例1
115.(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水,600克油酸混合均匀得到混合液b。
[0116]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0117]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0118]
(4)取100克所得前驱体、5克羟丙基纤维素(造孔剂)、40克sio2、1.6g石墨(润滑剂、造孔剂)、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0119]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前述催化剂评价方法对所得催化剂进行测评,其结果如表3所示。
[0120]
实施例2
[0121]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克1-丙醇、100克去离子水,100克硬脂酸混合均匀得到混合液b。
[0122]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0123]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0124]
(4)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0125]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前述催化剂评价方法对所得催化剂进行测评,其结果如表3所示。
[0126]
实施例3
[0127]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水,600克油酸混合均匀得到混合液b。
[0128]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0129]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0130]
(4)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0131]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前述催化剂评价方法对所得催化剂进行测评,其结果如表3所示。
[0132]
实施例4
[0133]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])、和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水,600克月桂酸混合均匀得到混合液b。
[0134]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0135]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0136]
(4)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0137]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0138]
实施例5
[0139]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])、和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克甲醇、100克去离子水,600克油酸混合均匀得到混合液b。
[0140]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0141]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0142]
(4)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0143]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0144]
实施例6
[0145]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])、和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水,600克油酸混合均匀得到混合液b。
[0146]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加5wt%的naoh水溶液,至ph=7值。
[0147]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0148]
(4)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0149]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0150]
实施例7
[0151]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])、和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水,600克油酸混合均匀得到混合液b。
[0152]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0153]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0154]
(4)取100克所得前驱体、15克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0155]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如
表3所示。
[0156]
实施例8
[0157]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])、和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水,600克油酸混合均匀得到混合液b。
[0158]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0159]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0160]
(4)取100克所得前驱体、25克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0161]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0162]
实施例9
[0163]
按照实施例1的方法,不同的是,使用50g乙醇和50g1-丙醇代替100g乙醇,xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0164]
实施例10
[0165]
按照实施例1的方法,不同的是,使用400g油酸和200g硬脂酸代替600g油酸,xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0166]
实施例11
[0167]
按照实施例1的方法,不同的是,8.55克硝酸铜(cu(no3)2·
3h2o)和10.32克ni(no3)2·
6h2o代替17.1克硝酸铜。xrd图谱在2θ=31.5
°
,2θ=28.2
°
,2θ=24.9
°
,2θ=23.3
°
,2θ=22.2
°
等处均有特征峰,xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0168]
对比例1
[0169]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])、和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a,滴加25wt%的氨水溶液,至ph=7值。
[0170]
(2)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0171]
(3)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0172]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果
如表3所示。
[0173]
对比例2
[0174]
(1)将22.0克偏钒酸铵(nh4vo3)、100.0克七钼酸铵((nh4)6mo7o
24
·
4h2o)、38.2克钨酸铵((nh4)5h5[h2(wo4)6])和17.1克硝酸铜(cu(no3)2·
3h2o)溶解于300ml去离子水中,得到混合液a。将100克乙醇、100克去离子水混合均匀得到混合液b。
[0175]
(2)混合液a滴加至不断搅拌的混合液b中,再继续滴加25wt%的氨水,至ph=7值。
[0176]
(3)将得到的悬浊液转移至烘箱中,80℃烘干48h后进行粉碎,得到催化剂前驱体。
[0177]
(4)取100克所得前驱体、5克羟丙基纤维素、40克sio2、1.6g石墨、3.2g去离子水混合均匀,通过压片机设置压片压力3kn,压片成型得到直径为5mm,片厚4mm的圆形片状成型催化剂,最后在385℃焙烧20小时,即得到丙烯酸合成用催化剂。
[0178]
通过氮气吸脱附测试所得催化剂的孔结构,如表1所示。xrd测试的2θ=24.9与2θ=22.2的峰强度比值如表2所示。通过前催化剂述评价方法对所得催化剂进行测评,其结果如表3所示。
[0179]
表1:催化剂总孔容和比例
[0180]
实施例总孔容(cm3/g)2-50nm孔比例50-1000nm孔比例10.05168%32%20.02745%55%30.04965%35%40.04159%41%50.03553%47&60.01933%67%70.05563%37%80.06058%42%90.05366%34%100.05163%37%110.05065%35%对比例10.00821%79%对比例20.00923%77%
[0181]
表2:催化剂xrd测试2θ=24.9和2θ=22.2强度值和比值
[0182]
实施例2θ=24.9峰强度2θ=22.2峰强度比值基线强度121414550.12733.6239814520.26124.9322514580.13631.6435514600.21625.6545614560.29928.8656314580.37330.0721414550.12733.6821414550.12733.6921214530.12731.8
1021314540.12732.41122715420.13030.4对比例180514550.54816.3对比例280514550.54816.3
[0183]
表3:催化剂活性测试性能
[0184][0185][0186]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
[0187]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。