1.本发明涉及生物化学技术领域,具体为一种自身可发光的磁性微球的制备方法及其应用。
背景技术:
2.在目前的实际应用中,针对化学发光分析仪的质控主要有两种方法。第一种方法:由于市面上现有的磁性微球本身不具有发光值,所以对发光分析仪进行质控时需结合发光免疫分析检测试剂盒使用。不同厂家试剂盒主要组分的生产包括包被反应板、标记物制备、各种溶液的配置、冻干、分包装等步骤,使用的原料不同,生产过程也有一定的差异,因而使用不同厂家的发光免疫分析试剂盒进行仪器质控时产生的不可控因素也会较多,对发光仪的质控效果不同无法得出确切的结论,可信度不足。另外,这一方法由于发光免疫分析试剂盒的使用,无疑增加了额外的成本,也成为这一方法的缺点之一。第二种方法:用一种标准光源对发光分析仪的测量室进行检测进而达到对测量室质控的目的。然而上文提到,化学发光免疫分析仪不仅包括试测量室检测区,还有试剂区、样品区、反应测试管加样区、反应废液区等很多不同的区域,显然这种方法是很局限的:它无法实现仪器其他部分的质控即不能满足化学发光分析仪整体的质控,功能单一,效果片面,结果不具有足够的说服力。市面上现有的普通磁性微球和化学发光分析仪的质控方法无法应对更为复杂的应用要求,不可避免地受到一定限制。
技术实现要素:
3.本发明的目的在于提供一种自身可发光的磁性微球的制备方法及其应用,以解决上述背景技术中提出的问题。
4.为了解决上述技术问题,本发明提供如下技术方案:一种自身可发光的磁性微球,具有以下特征:所述自身可发光的磁性微球为磁性微球表面活化后,在其表面直接或间接偶联包被发光物质后制得;
5.其中,所述发光物质为吖啶酰肼或吖啶酯中的任意一种。
6.一种自身可发光的磁性微球的制备方法,具有以下特征:所述自身可发光的磁性微球的制备方法包括直接法与间接偶联法;
7.其中直接法包括以下步骤:
8.s1.制备羧基化磁性微球;
9.s2.清洗羧基化磁性微球表面,并使用碳二亚胺活化;
10.s3.将活化后的羧基化磁性微球与吖啶酰肼溶液混合,加入氨水与硅酸四乙酯振荡反应,收集反应后磁性微球并清洗表面,得到自身可发光的磁性微球;
11.其中,间接偶联法包括以下步骤:
12.a.制备磁性微球;
13.b.将牛血清蛋白与cb缓冲液混合,配置为牛血清蛋白溶液;
14.c.向牛血清蛋白溶液中加入生物素、吖啶酯与cb缓冲液,震荡反应,加入tris终止液终止反应,对其进行透析并收集透析液备用;
15.d.将步骤a制备的磁性微球与步骤c制备的透析液混合反应,反应结束后收集磁性微球,并洗涤,得到自身可发光微球。
16.进一步的,直接法制备自身可发光的磁性微球,具体包括以下步骤:
17.s1.将fecl3.6h2o、fecl2.4h2o与纯水混合,搅拌均匀,调节溶液ph值为10-11,水浴升温至55-65℃,搅拌反应1-2h,反应结束后滴加油酸,升温至80-90℃,继续反应0.5-1h,对反应产物磁分离,得到羧基化磁性微球;
18.s2.将羧基化磁性微球与mes溶液混合,振荡10-20min,磁分离混合溶液,收集羧基化磁性微球,重复3-4次后,将羧基化磁性微球与碳二亚胺混合均匀,室温震荡处理30-45min后,得到活化羧基磁性微球;
19.s3.将活化羧基磁性微球与c-b08缓冲溶液混合配置为磁珠溶液,将吖啶酰肼加入至环己烷中,避光条件下超声分散10-15min,配置为吖啶酰肼溶液,并将其滴加至磁珠溶液内,并立即加入氨水,充分搅拌反应2-3h,滴加硅酸四乙酯,滴加时间为4-6h,滴加结束后,收集磁性微球,并使用无水乙醇与去离子水交替洗涤3-5次,真空干燥2-4h后,得到自身可发光的磁性微球。
20.进一步的,按重量份数计,步骤s1中,fecl3.6h2o、fecl2.4h2o、纯水与油酸的比例为(1-2g):(4-5g):(70-75ml):(2-3ml);
21.步骤s1中,油酸的滴加时间为2-3min。
22.进一步的,步骤s3中,所述磁珠溶液、吖啶酰肼溶液、氨水与硅酸四乙酯的体积比为(30-40):(8-10):(16-20):(0.15-0.2)。
23.进一步的,步骤s3中,所述磁珠溶液的浓度为2.5-3.0mg/ml;所述吖啶酰肼溶液的浓度为1-1.5ul/ml;所述氨水的浓度为2%-2.5%。
24.进一步的,步骤s3中,所述c-b08缓冲液,ph值为7.3-7.5,含有45-55mmtris、0.8-1%nacl、0.08-0.12%p300、0.8-1.2%bsa、0.14-0.16%tw-20与4-6%甘油,余量为水。
25.本发明首先采用了直接在磁性微球表面包覆发光物质的方法制备了自身可发光的磁性微球,首先使用铁质原料合成微小的磁性微球,之后将其与油酸混合反应,在其表面生成具有较强电负性的羧基基团,羧基基团会与吖啶酰肼之间生成氢键,从而使得吖啶酰肼可以包覆在磁性微球的表面,达到自身可发光的目的。
26.进一步的,间接偶联法制备自身可发光的磁性微球,具体包括以下步骤:
27.a.将fecl3.6h2o溶于乙二醇中,搅拌至澄清后,水浴升温至40-50℃,加入peg4000,继续搅拌30-45min,加入三水合乙酸钠,搅拌至完全溶解后,升温至200-220℃密闭反应8-12h,冷却至室温后收集反应产物,使用无水乙醇与去离子水交替洗涤反应产物3-5次,并真空干燥后得到磁性微球;
28.b.将牛血清蛋白与cb缓冲液混合,配制为牛血清蛋白溶液;
29.c.向牛血清蛋白溶液中加入生物素、吖啶酯与cb缓冲液,震荡均匀后,水浴升温至36.5-37.5℃反应60-80min,反应结束后加入tris终止液,混合均匀后,对其进行透析,直至透析液发光值降至9000-10000,停止透析并收集透析液;
30.d.将步骤a制备的磁性微球分散至环己烷中,并使用c-b08缓冲溶液稀释,得到磁
珠混合溶液,将其与步骤c制备的透析液混合均匀,37℃摇床反应2-3h,摇床转速为100-150rpm,反应结束后收集磁性微球,重复上述步骤3-5次,并使用无水乙醇与去离子水洗涤,得到自身可发光微球。
31.进一步的,步骤a中,所述fecl3.6h2o、peg4000、三水合乙酸钠的质量比为(1.1-1.5):(0.8-1.2):(3-4);
32.步骤b中,牛血清蛋白溶液的浓度为4.5-5g/100ml。
33.进一步的,步骤c中,牛血清蛋白溶液、生物素、吖啶酯、cb缓冲液与tris终止液的体积比为(40-50):(0.32-0.4):(4.4-5.5):(360-450):(48-60);
34.步骤d中,磁珠混合溶液与透析液的体积比为(0.8-1):1。
35.进一步的,所述自身可发光的磁性微球可应用于全自动化学发光免疫分析仪的质控。
36.与现有技术相比,本发明所达到的有益效果是:本发明制备的可自身发光的磁性微球,首先对磁性微球表面进行活化处理,在其表面生成具有强电负性的羧基,之后又将化学发光物质通过直接或间接的方式与磁性微球混合,与其表面的羧基形成氢键,最终形成牢固氢键将发光物质与磁性微球连接,制备出可自身发光的磁性微球。本发明制备的磁性微球本身自带发光值,可以减少发光仪检测过程中的误差,达到增强其检测效果稳定的目的。
具体实施方式
37.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
38.实施例1.
39.一种自身可发光的磁性微球的制备方法,包括以下步骤:
40.s1.将1gfecl3.6h2o、4g fecl2.4h2o与72ml纯水混合,搅拌均匀,调节溶液ph值为10,水浴升温至55℃,搅拌反应1h,反应结束后滴加油酸2ml,升温至80℃,继续反应0.5h,对反应产物磁分离,得到羧基化磁性微球;
41.s2.将羧基化磁性微球与mes溶液混合,置于涡旋仪上振荡10min,磁分离混合溶液,收集羧基化磁性微球,重复3次后,将羧基化磁性微球与碳二亚胺混合均匀,室温震荡处理30-45min后,得到活化羧基磁性微球;
42.s3.将活化羧基磁性微球与c-b08缓冲溶液混合配置为30ml浓度为2.5mg/ml的磁珠溶液,量取10ul吖啶酰肼加入至10ml环己烷中,避光条件下超声分散10min,配置为吖啶酰肼溶液,并将其滴加至磁珠溶液内,滴加结束后立即加入20ml浓度为2%的氨水,充分搅拌反应2h,滴加0.15ml硅酸四乙酯,滴加时间为4h,滴加结束后,使用外部磁铁收集磁性微球,并使用无水乙醇与去离子水交替洗涤3次,真空60℃干燥2h后,得到自身可发光的磁性微球;
43.其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
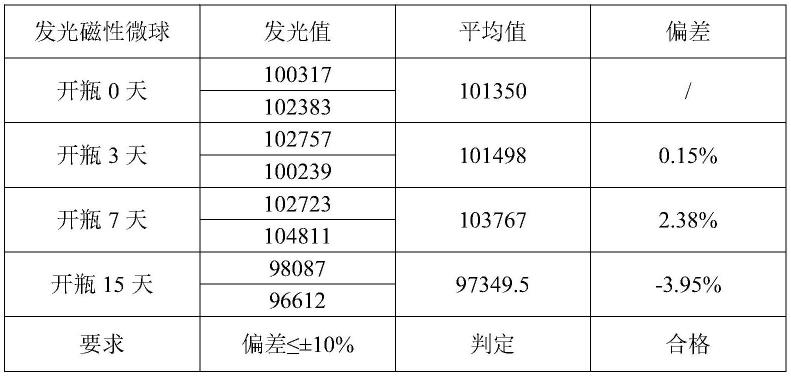
44.s4.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表1:实施例1发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于4%。
45.表1.实施例1发光磁珠开瓶稳定性结果表
[0046][0047]
s5.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表2:实施例1发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于3%。
[0048]
表2.实施例1发光磁性微球加速稳定性结果表
[0049][0050]
s6.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表3:实施例1发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于2%。
[0051]
表3.实施例1发光磁性微球冻融稳定性结果表
[0052][0053]
s7.对发光磁性微球进行重复性检测:将发光磁性微球在同样条件下重复测定10次,计算10次测量结果的平均值m和标准差sd,得出变异系数cv,结果应≤8%。检测结果如下表4:实施例1发光磁性微球重复性结果表所示。结果表明本发明的发光磁性微球cv为2%,小于8%,差异较小,重复性很好。
[0054]
表4.实施例1发光磁性微球重复性结果表
[0055]
序号发光值1100672210113539725549968259810161006907105101810048891039531099686av100676sd2377.38cv2%要求偏差≤
±
8%判定合格
[0056]
实施例2.
[0057]
与实施例1相比,本实施例增加了步骤s1中油酸的添加量;
[0058]
一种自身可发光的磁性微球的制备方法,包括以下步骤:
[0059]
s1.将1g fecl3.6h2o、4g fecl2.4h2o与72ml纯水混合,搅拌均匀,调节溶液ph值为10,水浴升温至55℃,搅拌反应1h,反应结束后滴加油酸3ml,升温至80℃,继续反应0.5h,对反应产物磁分离,得到羧基化磁性微球;
[0060]
s2.将羧基化磁性微球与mes溶液混合,置于涡旋仪上振荡10min,磁分离混合溶
液,收集羧基化磁性微球,重复3次后,将羧基化磁性微球与碳二亚胺混合均匀,室温震荡处理30-45min后,得到活化羧基磁性微球;
[0061]
s3.将活化羧基磁性微球与c-b08缓冲溶液混合配置为30ml浓度为2.5mg/ml的磁珠溶液,量取10ul吖啶酰肼加入至10ml环己烷中,避光条件下超声分散10min,配置为吖啶酰肼溶液,并将其滴加至磁珠溶液内,滴加结束后立即加入20ml浓度为2%的氨水,充分搅拌反应2h,滴加0.15ml硅酸四乙酯,滴加时间为4h,滴加结束后,使用外部磁铁收集磁性微球,并使用无水乙醇与去离子水交替洗涤3次,真空60℃干燥2h后,得到自身可发光的磁性微球;
[0062]
其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
[0063]
s4.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表5:实施例2发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于3%。
[0064]
表5.实施例2发光磁珠开瓶稳定性结果表
[0065][0066][0067]
s5.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表6:实施例2发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于3%。
[0068]
表6.实施例2发光磁性微球加速稳定性结果表
[0069][0070]
s6.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表7:实施例2发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于4%。
[0071]
表7.实施例2发光磁性微球冻融稳定性结果表
[0072][0073][0074]
s7.对发光磁性微球进行重复性检测:将发光磁性微球在同样条件下重复测定10次,计算10次测量结果的平均值m和标准差sd,得出变异系数cv,结果应≤8%。检测结果如下表8:实施例2发光磁性微球重复性结果表所示。结果表明本发明的发光磁性微球cv为3%,小于8%,差异较小,重复性很好。
[0075]
表8.实施例2发光磁性微球重复性结果表
[0076]
序号发光值11006722984483102369
497154598315610482079900789901591035761099269av100265sd2521.52cv3%要求偏差≤
±
8%判定合格
[0077]
实施例3.
[0078]
与实施例1相比,本实施例增加了步骤s3中吖啶酰肼的添加量;
[0079]
一种自身可发光的磁性微球的制备方法,包括以下步骤:
[0080]
s1.将1g fecl3.6h2o、4g fecl2.4h2o与72ml纯水混合,搅拌均匀,调节溶液ph值为10,水浴升温至55℃,搅拌反应1h,反应结束后滴加油酸2ml,升温至80℃,继续反应0.5h,对反应产物磁分离,得到羧基化磁性微球;
[0081]
s2.将羧基化磁性微球与mes溶液混合,置于涡旋仪上振荡10min,磁分离混合溶液,收集羧基化磁性微球,重复3次后,将羧基化磁性微球与碳二亚胺混合均匀,室温震荡处理30-45min后,得到活化羧基磁性微球;
[0082]
s3.将活化羧基磁性微球与c-b08缓冲溶液混合配置为30ml浓度为2.5mg/ml的磁珠溶液,量取15ul吖啶酰肼加入至10ml环己烷中,避光条件下超声分散10min,配置为吖啶酰肼溶液,并将其滴加至磁珠溶液内,滴加结束后立即加入20ml浓度为2%的氨水,充分搅拌反应2h,滴加0.15ml硅酸四乙酯,滴加时间为4h,滴加结束后,使用外部磁铁收集磁性微球,并使用无水乙醇与去离子水交替洗涤3次,真空60℃干燥2h后,得到自身可发光的磁性微球。
[0083]
其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
[0084]
s4.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表9:实施例3发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于2%。
[0085]
表9.实施例3发光磁珠开瓶稳定性结果表
[0086][0087]
s5.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表10:实施例3发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于4%。
[0088]
表10.实施例3发光磁性微球加速稳定性结果表
[0089][0090][0091]
s6.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表11:实施例3发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于3%。
[0092]
表11.实施例3发光磁性微球冻融稳定性结果表
cb缓冲液,震荡均匀后,水浴升温至37℃反应60min,反应结束后加入48ul tris终止液,混合均匀后,对其进行透析,直至透析液发光值降至9000-10000,停止透析并收集透析液;
[0103]
d.量取10mg步骤a制备的磁性微球分散至10ml环己烷中,并使用c-b08缓冲溶液稀释,得到浓度为2.5mg/ml的磁珠混合溶液,将其与步骤c制备的透析液按照体积比为0.8:1的比例混合均匀,37℃摇床反应2h,摇床转速为100rpm,反应结束后使用磁铁收集磁性微球,重复上述步骤3次,之后使用无水乙醇与去离子水洗涤,得到自身可发光微球;
[0104]
其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
[0105]
e.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表13:实施例4发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于2%。
[0106]
表13.实施例4发光磁珠开瓶稳定性结果表
[0107][0108]
f.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表14:实施例4发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于6%。
[0109]
表14.实施例4发光磁性微球加速稳定性结果表
[0110][0111]
g.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表15:实施例4发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于5%。
[0112]
表15.实施例4发光磁性微球冻融稳定性结果表
[0113][0114][0115]
h.对发光磁性微球进行重复性检测:将发光磁性微球在同样条件下重复测定10次,计算10次测量结果的平均值m和标准差sd,得出变异系数cv,结果应≤8%。检测结果如下表16:实施例4发光磁性微球重复性结果表所示。结果表明本发明的发光磁性微球cv为3%,小于8%,差异较小,重复性很好。
[0116]
表16.实施例4发光磁性微球重复性结果表
[0117]
序号发光值115468021569673148190
41534825148876615635271446808146711915465810143783av150838sd4943.01cv3%要求偏差≤
±
8%判定合格
[0118]
实施例5.
[0119]
与实施例4相比,本实施例增加了步骤d中磁珠混合溶液与透析液的混合比例;
[0120]
一种自身可发光的磁性微球的制备方法,包括以下步骤:
[0121]
a.将1.1g fecl3.6h2o溶于40ml乙二醇中,搅拌至澄清后,水浴升温至40℃,加入0.8g peg4000,继续搅拌30min,加入3g三水合乙酸钠,搅拌至完全溶解后,将其转移至高温高压反应釜中,马弗炉升温至200℃,密闭反应8h,冷却至室温后使用磁铁收集反应产物,使用无水乙醇与去离子水交替洗涤反应产物3次,并真空60℃干燥2h后得到磁性微球;
[0122]
b.将4.5g牛血清蛋白与100ml cb缓冲液混合,配置为牛血清蛋白溶液;
[0123]
c.量取40ul牛血清蛋白溶液,并向其中加入0.32ul生物素、4.4ul吖啶酯与360ul cb缓冲液,震荡均匀后,水浴升温至37℃反应60min,反应结束后加入48ul tris终止液,混合均匀后,对其进行透析,直至透析液发光值降至9000-10000,停止透析并收集透析液;
[0124]
d.量取10mg步骤a制备的磁性微球分散至10ml环己烷中,并使用c-b08缓冲溶液稀释,得到浓度为2.5mg/ml的磁珠混合溶液,将其与步骤c制备的透析液按照体积比为1:1的比例混合均匀,37℃摇床反应2h,摇床转速为100rpm,反应结束后使用磁铁收集磁性微球,重复上述步骤3次,之后使用无水乙醇与去离子水洗涤,得到自身可发光微球;
[0125]
其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
[0126]
e.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表17:实施例5发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于4%。
[0127]
表17.实施例5发光磁珠开瓶稳定性结果表
[0128][0129]
f.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表18:实施例5发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于5%。
[0130]
表18.实施例5发光磁性微球加速稳定性结果表
[0131][0132]
g.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表19:实施例5发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于6%。
[0133]
表19.实施例5发光磁性微球冻融稳定性结果表
[0134][0135]
h.对发光磁性微球进行重复性检测:将发光磁性微球在同样条件下重复测定10次,计算10次测量结果的平均值m和标准差sd,得出变异系数cv,结果应≤8%。检测结果如下表20:实施例5发光磁性微球重复性结果表所示。结果表明本发明的发光磁性微球cv为3%,小于8%,差异较小,重复性很好。
[0136]
表20.实施例5发光磁性微球重复性结果表
[0137]
序号发光值1985322100183397746498102510321861046217104657810442691030091098794av101329sd2919.46cv3%要求偏差≤
±
8%判定合格
[0138]
实施例6.
[0139]
一种自身可发光的磁性微球的制备方法,包括以下步骤:
[0140]
a.将1.35g fecl3.6h2o溶于40ml乙二醇中,搅拌至澄清后,水浴升温至50℃,加入1g peg4000,继续搅拌30min,加入3.6g三水合乙酸钠,搅拌至完全溶解后,将其转移至高温高压反应釜中,马弗炉升温至200℃,密闭反应8h,冷却至室温后使用磁铁收集反应产物,使用无水乙醇与去离子水交替洗涤反应产物3次,并真空60℃干燥2h后得到磁性微球;
[0141]
b.将5g牛血清蛋白与100ml cb缓冲液混合,配置为牛血清蛋白溶液;
[0142]
c.量取50ul牛血清蛋白溶液,并向其中加入0.4ul生物素、5.5ul吖啶酯与450ul cb缓冲液,震荡均匀后,水浴升温至37℃反应60min,反应结束后加入60ul tris终止液,混合均匀后,对其进行透析,直至透析液发光值降至9000-10000,停止透析并收集透析液;
[0143]
d.量取10mg步骤a制备的磁性微球分散至10ml环己烷中,并使用c-b08缓冲溶液稀释,得到浓度为2.5mg/ml的磁珠混合溶液,将其与步骤c制备的透析液按照体积比为1:1的比例混合均匀,37℃摇床反应2h,摇床转速为100rpm,反应结束后使用磁铁收集磁性微球,重复上述步骤5次,之后使用无水乙醇与去离子水洗涤,得到自身可发光微球;
[0144]
其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
[0145]
e.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表21:实施例6发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于4%。
[0146]
表21.实施例6发光磁珠开瓶稳定性结果表
[0147][0148]
f.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表22:实施例6发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于2%。
[0149]
表22.实施例6发光磁性微球加速稳定性结果表
[0150][0151]
g.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表23:实施例6发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于4%。
[0152]
表23.实施例6发光磁性微球冻融稳定性结果表
[0153][0154]
h.对发光磁性微球进行重复性检测:将发光磁性微球在同样条件下重复测定10次,计算10次测量结果的平均值m和标准差sd,得出变异系数cv,结果应≤8%。检测结果如下表24:实施例6发光磁性微球重复性结果表所示。结果表明本发明的发光磁性微球cv为3%,小于8%,差异较小,重复性很好。
[0155]
表24.实施例6发光磁性微球重复性结果表
[0156]
序号发光值11052072985403980924100533
5996776105050796741810352891020191099355av100874sd2956.98cv3%要求偏差≤
±
8%判定合格
[0157]
实施例7.
[0158]
一种自身可发光的磁性微球的制备方法,包括以下步骤:
[0159]
a.将1.5g fecl3.6h2o溶于40ml乙二醇中,搅拌至澄清后,水浴升温至50℃,加入1.2g peg4000,继续搅拌30min,加入4g三水合乙酸钠,搅拌至完全溶解后,将其转移至高温高压反应釜中,马弗炉升温至220℃,密闭反应12h,冷却至室温后使用磁铁收集反应产物,使用无水乙醇与去离子水交替洗涤反应产物5次,并真空60℃干燥2h后得到磁性微球;
[0160]
b.将5g牛血清蛋白与100ml cb缓冲液混合,配置为牛血清蛋白溶液;
[0161]
c.量取50ul牛血清蛋白溶液,并向其中加入0.4ul生物素、5.5ul吖啶酯与450ul cb缓冲液,震荡均匀后,水浴升温至37℃反应60min,反应结束后加入60ul tris终止液,混合均匀后,对其进行透析,直至透析液发光值降至9000-10000,停止透析并收集透析液;
[0162]
d.量取10mg步骤a制备的磁性微球分散至10ml环己烷中,并使用c-b08缓冲溶液稀释,得到浓度为2.5mg/ml的磁珠混合溶液,将其与步骤c制备的透析液按照体积比为1:1的比例混合均匀,37℃摇床反应2h,摇床转速为150rpm,反应结束后使用磁铁收集磁性微球,重复上述步骤5次,之后使用无水乙醇与去离子水洗涤,得到自身可发光微球。
[0163]
其中所述c-b08缓冲液ph为7.4,含有50mmtris、0.9%nacl、0.1%p300、1%bsa、0.15%tw-20与5%甘油,余量为水;
[0164]
e.对发光磁性微球进行开瓶稳定性检测:将发光磁性微球于2~8℃开瓶储存,分别于第0天、第3天、第7天、第15天各取样10ul检测发光值,与开瓶0天的磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表25:实施例7发光磁性微球开瓶稳定性结果表所示。从结果中可以看出,本发明的发光磁珠开瓶非常稳定,偏差小于4%。
[0165]
表25.实施例7发光磁珠开瓶稳定性结果表
[0166][0167]
f.对发光磁珠进行加速稳定性检测:将发光磁性微球于37℃储存,分别于第1天、第3天、第5天、第7天各取样10ul检测发光值,与未加速的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表26:实施例7发光磁性微球加速稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球加速后仍然非常稳定,偏差小于3%。
[0168]
表26.实施例7发光磁性微球加速稳定性结果表
[0169][0170]
g.对发光磁性微球进行冻融稳定性检测:将发光磁性微球置于-20℃环境冻融,冻融1次、冻融2次、冻融3次,每次冻融时间在24h以上,复融时间不低于48h,冻融完成后与未冻融的发光磁性微球相比偏差,其偏差应在
±
10%以内。检测发光值如下表27:实施例7发光磁性微球冻融稳定性结果表所示。从结果中可以看出,本发明的发光磁性微球冻融非常稳定,偏差小于3%。
[0171]
表27.实施例7发光磁性微球冻融稳定性结果表
[0172][0173]
h.对发光磁性微球进行重复性检测:将发光磁性微球在同样条件下重复测定10次,计算10次测量结果的平均值m和标准差sd,得出变异系数cv,结果应≤8%。检测结果如下表28:实施例7发光磁性微球重复性结果表所示。结果表明本发明的发光磁性微球cv为3%,小于8%,差异较小,重复性很好。
[0174]
表28.实施例7发光磁性微球重复性结果表
[0175]
序号发光值1100452210496931046694966295101389610070779991689959099599510100088av100440sd2882.34cv3%要求偏差≤
±
8%判定合格
[0176]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。