1.本发明涉及微流控技术领域,具体涉及一种氟油组合物及其制备方法。
背景技术:
2.目前在体外诊断、纳米材料制备及环境监测等领域都对微滴精确操作有着广泛的需求。近些年来,在文献中报道了许多种微滴生成技术,如膜乳化法、喷雾乳化法、微流控芯片法等。其中微流控芯片法作为目前较新的微滴生成技术,在微滴生成操作方面以及耗材成本控制方面均具有较好的前景。
3.微滴式数字pcr(droplet digital pcr,ddpcr)是一种基于单分子pcr方法来计数的核酸定量方法,主要采用微流控技术将其样品进行微滴化处理,形成几万个纳升级别微滴,每个微滴或不含核酸靶分子或含有一个或数个核酸靶分子。经过pcr扩增之后,对微滴进行逐个计数检测,最后根据泊松分布原理及阳性微滴数目即可得到核酸的拷贝数和浓度。目前为了使微滴经过pcr热循环之后依旧保持原形貌,需要热稳定性高的生成油作为微滴的油相。比较常用的油相为氟油,例如:hfe-7500、hfe-7100、fc-40,搭配使用的表面活性剂有pfpe-peg-pfpe、abil
tm em90、1,1,2,2-四氢全氟癸醇等,但是现有用于氟油的表面活性剂往往只能应用于常温环境,在pcr热循环后无法保持其表面性能。
4.中国专利文献cn106823990a公开了一种用于制备微滴式数字pcr中液滴的油相组合物,其采用矿物油、硅油、、十二烷、十四烷、十六烷、和十八烷其中一种作为油相,表面活性剂采用全氟聚醚接枝壳聚糖,该专利合成路线简单产率显著提高,纯化简单,成本低廉,但是使用该表面活性剂时会额外添加防蒸发剂,说明该表面活性剂热稳定性不高。同时,该反应涉及能污染环境的二氯亚砜等有毒试剂,环境危害较大。
5.中国专利文献cn106622017a公开了一种氟表面活性剂制备方法,表面活性剂主要是全氟聚醚接枝三羟甲基氨基甲烷,形成两嵌段共聚物。该专利制备简单,性能优越,一步法制备。其反应试剂主要采用三氟甲苯、四氢呋喃、二甲基亚砜中的一种,上述溶剂毒性大,对人体危害较为严重。
技术实现要素:
6.因此,本发明要解决的技术问题在于现有氟油微滴的成本,毒性大,重复性差的问题,从而提供一种氟油组合物及其制备方法。
7.为此,本发明采用如下技术方案:
8.本发明提供一种氟油组合物,包括氟油和表面活性剂,所述表面活性剂包括结构如式i和/或式ii的化合物:
[0009][0010][0011]
其中n为20-80。
[0012]
优选地,所述表面活性剂由结构如式i和式ii的化合物组成,所述式i和式ii的化合物的摩尔比为1:2。
[0013]
进一步地,以质量百分比计,所述表面活性剂占所述的氟油组合物的0.5%-2%;
[0014]
所述氟油组合物中的氟油包括hfe-7500和1h,1h,2h,2h-全氟-1-辛醇,hfe-7500与1h,1h,2h,2h-全氟-1-辛醇的体积比为1000:1。
[0015]
本发明还提供上述氟油组合物的制备方法,包括如下步骤:
[0016]
s1:将式iii所示的全氟聚醚与草酰氯反应生成式iv所示的全氟聚醚酰氯;
[0017]
s2:式iv所示的全氟聚醚酰氯与己二酸二酰肼反应生成式i和/或式ii所示的化合物;
[0018]
s3:使用氢氧化钠溶液和盐酸对得到的式i和/或式ii的化合物交替洗涤,干燥后得到表面活性剂;
[0019]
s4:将得到的表面活性剂和氟油混合,形成所述氟油组合物;
[0020]
其中,式iii为
[0021]
式iv为
[0022]
进一步地,步骤s1中,反应条件在氮气条件下进行,反应温度为60-80℃,反应时间为12-24h,全氟聚醚与草酰氯的质量比为0.1-0.5:1;
[0023]
所述全氟聚醚的平均分子量为2500-7000;
[0024]
优选,采用krytox公司型号为157fsl的全氟聚醚。
[0025]
步骤s2中,全氟聚醚酰氯与溶于二氯甲烷的己二酸二酰肼在60-75℃下进行反应,生成全氟聚醚-己二酸二酰肼嵌段共聚物。
[0026]
优选地,全氟聚醚酰氯与己二酸二酰肼的摩尔比为1:0.5-1;
[0027]
二氯甲烷与己二酸二酰肼的质量比为20:0.1-0.174。。
[0028]
步骤s3中,氢氧化钠溶液的浓度为0.1m-1m,盐酸的浓度为0.1m-1m,洗涤次数为1-3次。
[0029]
步骤s1前还包括将全氟聚醚预处理的步骤,所述预处理为将全氟聚醚溶解在氟油hfe-7100中,加入分子筛,加热回流脱水;
[0030]
全氟聚醚与氟油hfe-7100的质量比为1:5-10,回流温度为60-75℃,回流时间为8-12h。
[0031]
步骤s2中,所述反应在氟油中进行,所述氟油为hfe-7500。
[0032]
本发明技术方案,具有如下优点:
[0033]
(1)本发明制得氟油组合物作为pcr微滴生成的油相使用时,可以实现在液滴式数字pcr上的应用,且微滴具有一定的热稳定性,具有优越的表面性能。
[0034]
(2)本发明制备过程中,使用hfe-7100可以避免草酰氯的水解,使用hfe-7500可以将最终产物溶解在其中,便于后期的纯化步骤。
[0035]
(3)本发明最后使用氢氧化钠溶液和盐酸交替洗涤的方式进行纯化,使用氢氧化钠保护了氨基,可以提高氟油的微滴数量和微滴的形貌以及荧光强度,再用盐酸洗涤中和剩余的氢氧化钠。
[0036]
(4)本发明优选限定式i和式ii结构的化合物的摩尔比为1:2。因为在式i结构的化合物时,其在油水界面上填充密度不高,如果和式ii的化合物联用之后,可以增大油水界面上的填充密度,使微滴更加稳定,能够经受pcr的高温热循环,而本发明优选限定的摩尔比是其最佳组成比例。
附图说明
[0037]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0038]
图1为本发明实施例1得到的表面活性剂的红外谱图;
[0039]
图2为本发明实施例1得到的表面活性剂的核磁共振谱图;
[0040]
图3为本发明试验例1中实施例1所生成的微滴在pcr热循环扩增前的显微镜图;
[0041]
图4为本发明试验例1中实施例1所生成的微滴在pcr热循环扩增后的显微镜图;
[0042]
图5为本发明试验例1中实施例1所生成的微滴在pcr热循环扩增后的数量显示图;
[0043]
图6为本发明试验例1中实施例1所生成的微滴在pcr热循环扩增后的扩增效率图。
具体实施方式
[0044]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0045]
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验
步骤的操作或条件即可进行。
[0046]
全氟聚醚(pfpe)购于krytox公司,型号为157fsl
[0047]
hfe-7500,hfe-7100均购于3m公司。
[0048]
其他试剂均为分析纯级别,购于sigma aldrich公司。
[0049]
实施例1
[0050]
本实施例提供一种氟油组合物:
[0051]
其中表面活性剂的合成路线如下:
[0052][0053]
具体制备方法如下:
[0054]
(1)取2.5g的式iii所示的pfpe加入20g的hfe-7100中,超声溶解3min,在65℃下回流12小时脱水。
[0055]
(2)加入10ml的草酰氯,在氮气氛围下,65℃下回流24h,得到式iv所示的全氟聚醚酰氯;将上述溶液在40℃下旋蒸10min,随后升高温度至70℃旋蒸10min,然后冷却至室温。
[0056]
(3)将0.13g的己二酸二酰肼加入20g的二氯甲烷中,超声溶解3min。
[0057]
(4)加入20g的hfe-7500溶解步骤(2)的产物,然后加入步骤(3)的混合液体,超声分散3min。
[0058]
(5)将步骤(4)所得的混合液体在氮气保护下在65℃下反应24h,得到式i和式ii所
示的化合物。
[0059]
(6)反应结束后,在90℃下旋蒸除去多余的hfe-7500。
[0060]
(7)使用0.1m的氢氧化钠和0.1m的盐酸溶液分别洗涤3次。
[0061]
(8)室温下真空干燥24h,得到表面活性剂。
[0062]
(9)将所得表面活性剂溶于氟油中,得到氟油组合物,所述表面活性剂为2wt%,所述氟油由hfe-7500和1h,1h,2h,2h-全氟-1-辛醇组成,hfe-7500与1h,1h,2h,2h-全氟-1-辛醇的体积比为1000:1。
[0063]
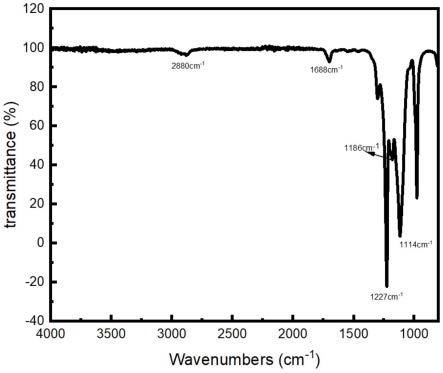
对步骤(8)所得表面活性剂进行傅里叶红外表征以及核磁共振表征,具体的表征图谱如图1和图2所示。如图1所示,在红外图谱中,位于1688cm-1
位置的特征峰对应于产物的酰胺-c=o双键,位于2889cm-1
、1227cm-1
和1186cm-1
位置的特征峰对应于产物的-ch2、-c-f和-c-n键。如图2的核磁图谱所示,δ6.0-6.5处的峰代表的是酰胺处连接的n-h的峰,δ3.0-3.75处的峰代表的是己二酸二酰肼处的亚甲基的峰。根据红外和核磁的图谱结果,即验证所得表面活性剂为式i和式ii结构的化合物组成:
[0064][0065]
其中n为20。
[0066]
其中式i和式ii结构的化合物的摩尔比为1:2。
[0067]
实施例2
[0068]
本实施例提供一种氟油组合物,和实施例1唯一的不同在于,己二酸二酰胺的加入量为0.174g,其中得到的表面活性剂中仅有式ii结构的化合物。
[0069]
实施例3
[0070]
本实施例提供一种氟油组合物,和实施例1唯一的不同在于,己二酸二酰胺的加入量为0.087g,其中得到的表面活性剂中仅有式i结构的化合物。
[0071]
实施例4
[0072]
本实施例提供一种氟油组合物,和实施例1唯一的不同在于,己二酸二酰胺的加入量为0.116g,其中得到的表面活性剂中式i和式ii结构的化合物的摩尔比为1:1。
[0073]
实施例5
[0074]
本实施例提供一种氟油组合物,和实施例1唯一的不同在于,己二酸二酰胺的加入量为0.104g,其中得到的表面活性剂中式i和式ii结构的化合物的摩尔比为2:1。
[0075]
试验例1
[0076]
将实施例1得到的氟油组合物作为pcr微滴生成油相。pcr微滴生成水相的制备方
法为将60μl的2x buffer(supermix,购于biorad公司)、10μl的正引物prime-f(10μm)、10μl的反向引物prime-r(10μm)、10μl的λdna(10ng/ml)、10ul的探针probe(10μm)混匀,滴加60μl超纯水混合均匀。
[0077]
取体积比为4:1的油相和水相,在微流控芯片中生成均一的性良好的油包水微滴(20μm-120μm),如图3所示。
[0078]
随后进行pcr热循环扩增实验。pcr热循环扩增前显微镜下观察到液滴的形貌如图3所示,pcr热循环扩增后显微镜下观察到微滴的形貌如图4所示。在显微镜下观察所示,实施例1制备的表活所生成的液滴经过pcr热循环30-50圈热循环后,没有看到破乳和融合的现象,大部分微滴均一性状态依旧保持良好,仍然呈现规律的蜂窝状排列,破乳及融合的微滴的cv值(变异系数)低于5%。
[0079]
将pcr热循环扩增的微滴进行上机检测(机器为biorad公司生产,型号:qx200),经过pcr热循环扩增之后微滴的个数如图5所示,微滴经过pcr热循环后,微滴的个数保持在13734个,说明大部分微滴没有破乳。如图6所示,可以看到经过pcr热扩增之后,有两条非常明显的条带,分别是阴性带和阳性带。说明微滴内部成功进行了扩增,表面活性剂并没有影响其扩增效率。
[0080]
对实施例2-5得到的氟油组合物做相同的测试,生成的微滴经过pcr热循环之后均出现了大部分破乳的现象,表面本技术实施例1特定配比的结构效果更好。
[0081]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。