1.本发明属于有机合成技术领域,尤其是指一种吡咯烷和四氢呋喃环化合物及其制备方法。
背景技术:
2.过渡金属催化的不对称环加成已经成为以高效和收敛的方式构建手性碳环或杂环和融合环系统的最有力的工具,已经大大简化了天然产物、药物和农用化学品的不对称合成。在过去的二十年里,harmata(j.am.chem.soc.2003,125,2058-2059),hsung(j.am.chem.soc.2005,127,50-51),uria&vicario(angew.chem.int.ed.2017,56,10535-10538)和jacobsen(science2017,358,761-764)在杂烯基阳离子催化不对称(3 4)环加成反应中取得了重大突破,呋喃是碳环的唯一受体。然而,杂烯基阳离子和2π配偶体之间存在不可逾越的鸿沟,其是阻止未催化的协同(3 2)途径的无匹配的前沿分子轨道。此外,钯-杂烯丙基转移过程与类似的钯-三甲基乙烷中间体的环加成明显不同(acc.chem.res.2020,53,1293-1305),并且已经广泛应用于不对称环加成。
3.为了弥补这一差距,紧接着报道了首先使用化学计量金属基试剂生成异烯丙基阳离子的有限数量的逐步(3 2)环加成反应(synthesis 2015,47,22-33)。最近,trost及其同事报道了令人印象深刻的催化产生和与富含电子的1,3-二烯的杂烯基阳离子的独特的(3 2)杂环加成,通过终止碳-氮或碳-氧键形成的途径产生吡咯烷和四氢呋喃,将杂烯基阳离子带入一个新的时代(science 2018,362,564-568,angew.chem.int.ed.2019,58,6396-6399)。然而,现有技术中涉及钯-杂烯丙基阳离子的杂环加成的不对称版本迄今尚未被探索。
技术实现要素:
4.为解决上述技术问题,本发明提供了一种手性吡咯烷和四氢呋喃环化合物及其制备方法。本发明的制备方法效率高、操作简单、反应条件温和、化学选择性和对映选择性优异。能对活性和药物分子衍生物进行修饰,具有较大的实施价值和应用前景。
5.本发明的第一个目的在于提供一种手性吡咯烷和四氢呋喃环化合物的制备方法,包括以下步骤:
[0006][0007]
化合物i或化合物ii和化合物iii在钯催化剂以及手性配体l*的存在下进行反应,得到手性吡咯烷和四氢呋喃环化合物ⅳ;
[0008][0009]
其中,x为o或nso2r,r为苯磺酰基、邻位甲基的苯磺酰基或对位甲基的苯磺酰基中的任意一种;
[0010]
中的虚线表示可以为环状的共轭二烯或非环状的共轭二烯。
[0011]
r1为甲基、乙基、正丁基、异丁基、苯基或苄基中的任意一种;
[0012]
r2为r
x
选自氢、卤素、甲基、正丁基、甲氧基、三氟甲基、三氟甲氧基或四甲基硅烷中的任意一种;
[0013]
r3为氢或甲基,且r2与r3不能同时为相同的基团。
[0014]
在本发明的一个实施例中,所述r2为噻吩基、呋喃基、1-乙烯基、环丙基、苄基、氧苄基、正丁基、1-苯乙炔基、1-丙基和叔丁基二甲基硅氧基、苄醚、苄酯或正己基中的任意一种。
[0015]
在本发明的一个实施例中,所述手性配体l*的化学结构式如下式所示:
[0016][0017]
其中,
[0018]
r4为氢或叔丁基;
[0019]
r5为氢、叔丁基、金刚烷基、3,5-二氟、3,5-二氯、5-氟、1,2,3,4,5-五氟、联苯、1-萘或2-萘中的任意一种;
[0020]
r6为氢或甲基。
[0021]
在本发明的一个实施例中,所述手性配体l*为下述化合物中的一种或多种:
[0022][0023]
在本发明的一个实施例中,所述钯催化剂为三(二亚苄基丙酮)二钯-氯仿加合物、三(二亚苄基丙酮)二钯、二-μ-氯双[(1,2,3-η)-1-苯基-2-丙烯-1-基]二钯、醋酸钯、氯化钯、三氟乙酸钯、二(乙酰丙酮)钯、氯化钯(π-肉桂基)二聚物、(1,5-环辛二烯)二氯化钯、四三苯基膦钯和三苯基膦二氯化钯中的一种或多种。
[0024]
在本发明的一个实施例中,所述化合物i或ii与所述化合物ⅲ摩尔比1:(1-5)。
[0025]
在本发明的一个实施例中,所述化合物i或ii与所述钯催化剂的摩尔比为1:(0.01-0.1)。
[0026]
在本发明的一个实施例中,所述钯催化剂与所述手性配体l*的摩尔比为1:(1-2)。
[0027]
在本发明的一个实施例中,反应条件为:惰性气氛中,在25℃-100℃下反应12h-96h。
[0028]
在本发明的一个实施例中,所述反应的反应溶剂为邻二甲苯、甲苯、四氢呋喃、乙腈、二氯甲烷、乙醚、1、4-二氧六环、甲醇、甲基叔丁基醚、异丙醚、正丁醚、乙二醇二甲醚或环戊基甲醚中的一种或多种。
[0029]
在本发明的一个实施例中,所述反应具体为:在惰性气氛下,反应溶剂b中,将所述钯催化剂与所述手性配体l*加入所述化合物i或化合物ii和化合物ⅲ中,并在25℃-100℃下搅拌12h-96h,反应结束后进行后处理,分离得到手性吡咯烷和四氢呋喃环化合物。
[0030]
在本发明的一个实施例中,所述惰性气氛中的气体是指在惰性气体和/或氮气。
[0031]
本发明的第二个目的在于提供一种由所述的制备方法所得手性吡咯烷和四氢呋喃环化合物,所述手性吡咯烷和四氢呋喃环化合物结构式如下所述:
[0032]
其中,x为o或nso2r,r为苯磺酰基、邻位甲基的苯磺酰基或对位甲基的苯磺酰基中的任意一种;
[0033]
r1为甲基、乙基、正丁基、异丁基、苯基或苄基中的任意一种;
[0034]
r2为r
x
选自氢、卤素、甲基、正丁基、甲氧基、三氟甲基、三氟甲氧基或四甲基硅烷中的任意一种;
[0035]
r3为氢或甲基,且r2与r3不能同时为相同的基团。
[0036]
进一步的,所述手性吡咯烷和四氢呋喃环化合物为2-((3ar,7as,e)-6-苯基-1-甲苯磺酰基-1,3a,4,5,7a-六氢-2h-吲哚-2-亚基)乙酸乙酯、乙基(e)-2-((s)-5-((e)-苯乙烯基)-1-对甲苯基吡咯烷-2-亚基)乙酸酯、
[0037]
[0038]
[0039][0040]
本发明的反应机理:杂烯丙基前体在钯与配体的作用下氧化加成之后,进行自发脱羧后,产生钯氨基烯丙基,随后,与共轭二烯中作用产生新的钯烯丙基物种,并且在钯烯丙基转移之后将进行碳杂键的还原消除以得到(3 2)环加成产物。
[0041]
本发明的上述技术方案相比现有技术具有以下优点:
[0042]
本发明所涉及的手性吡咯烷和四氢呋喃环化合物的制备方法,因为采用了一种合理设计的pc-phos配体因此在反应效率和选择性方面发挥了关键作用。实现了一种高度区域选择性、非对映选择性和对映选择性的钯催化的杂烯丙基前体与各种环状和非环状1,3-二烯的不对称杂环(3 2)加成反应,得到高度官能化的富含对映体的吡咯烷和四氢呋喃环化合物,具有效率高、操作简单、反应条件温和、化学选择性和对映选择性优异等特点,且能对活性或药物分子衍生物进行修饰,具有较大的实施价值和应用前景。
具体实施方式
[0043]
下面结合具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0044]
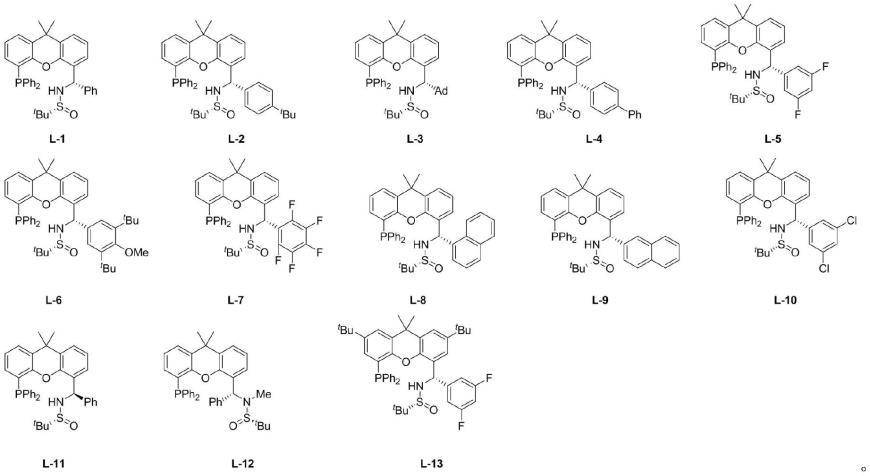
为了方便描述,下述实施例中使用的手性配体l*分别称之为l1-l13,各自对应的结构式如下所示:
[0045][0046]
上述手性配体l
*
的制备方法参考中国专利申请号为201710541779.1的制备方法进行,(专利中氧杂蒽骨架的单膦配体pc-phos即是本发明所述的手性配体l*),且未在该专利中公开的手性配体l
*
的表征如下:
[0047]
l-13:1h nmr(400mhz,cdcl3)δ9.06(s,1h),7.84(d,j=2.0hz,1h),7.55(d,j=2.0hz,1h),7.47
–
7.38(m,5h),7.32
–
7.26(m,6h),6.69
–
6.56(m,1h),1.70(s,3h),1.60(s,3h),1.32(s,9h),1.25(s,9h),1.15(s,9h);
13
c nmr(100mhz,cdcl3)δ
13
c nmr(101mhz,cdcl3)δ159.0,158.9,149.2,149.0,148.6,145.9,145.4,136.4,136.3,136.3,136.2,134.6,134.4,134.3,134.1,130.8,128.8,128.6,128.3,128.2,128.2,128.2,127.0,125.8,125.7,122.7,122.3,121.4,57.6,34.7,34.7,34.6,34.5,32.8,31.3,31.3,31.1,26.9,22.8;
31
pnmr(162mhz,cdcl3)δ16.7;
19
f nmr(376mhz,cdcl3)δ-109.1,-109.6;hrms(esi)calculated for[c
46h52
f2no2ps][m na]
:752.3497,found:752.3509;
[0048]
[α]
d20
=50.0(c=0.1,chcl3).
[0049]
《实施例1-16》
[0050]
2-((3ar,7as,e)-6-苯基-1-甲苯磺酰基-1,3a,4,5,7a-六氢-2h-吲哚-2-亚基)乙酸乙酯的制备方法
[0051]
本实施例1-16提供了2-((3ar,7as,e)-6-苯基-1-甲苯磺酰基-1,3a,4,5,7a-六氢-2h-吲哚-2-亚基)乙酸乙酯的制备方法。本实施例所用原料以及反应结果如表1所示,制备步骤如下:
[0052]
将手性配体l(0.024mmol),三(二亚苄基丙酮)二钯-氯仿加合物(10.4mg,0.01mmol),(z)-2-(2-氧代-3-甲苯噻唑啉-4-亚基)乙酸乙酯(65.0mg,0.2mmol)和2,3-二氢-1,1'-联苯(93.6mg,0.6mmol)加入反应瓶中,氮气氛围下加入溶剂(1.0ml),65℃下搅拌反应48h后,过柱分离制得2-((3ar,7as,e)-6-苯基-1-甲苯磺酰基-1,3a,4,5,7a-六氢-2h-吲哚-2-亚基)乙酸乙酯。
[0053]
表1实施例1-16
[0054][0055][0056]
《实施例17-33》
[0057]
乙基(e)-2-((s)-5-((e)-苯乙烯基)-1-对甲苯基吡咯烷-2-亚基)乙酸酯的制备
[0058]
本实施例17-33提供了一种乙基(e)-2-((s)-5-((e)-苯乙烯基)-1-对甲苯基吡咯烷-2-亚基)乙酸酯的制备方法,本实施例所用原料以及反应结果如表2所示,制备步骤如下:
[0059]
将手性配体l(0.024mmol),三(二亚苄基丙酮)二钯-氯仿加合物(10.4mg,0.01mmol),(z)-2-(2-氧代-3-甲苯噻唑啉-4-亚基)乙酸乙酯(65.0mg,0.2mmol)和(e)-丁-1,3-二烯-1-基苯(78.0mg,0.6mmol)加入反应瓶中,氮气氛围下加入溶剂(1.0ml),65℃下搅拌反应48h后,过柱分离制得乙基(e)-2-((s)-5-((e)-苯乙烯基)-1-对甲苯基吡咯烷-2-亚基)乙酸酯;
[0060]
表2实施例17-33
[0061]
[0062][0063]
《实施例34-91》
[0064]
底物的拓展
[0065]
本实施例对手性吡咯烷和四氢呋喃环化合物的制备方法所适应的底物进行了拓展,反应式为:
[0066][0067]
制备步骤如下:
[0068]
将手性配体l(0.024mmol),三(二亚苄基丙酮)二钯-氯仿加合物(10.4mg,0.01mmol),(z)-2-(2-氧代-3-甲苯噻唑啉-4-亚基)乙酸乙酯(0.2mmol)和2,3-二氢-1,1'-联苯(0.6mmol)加入反应瓶中,氮气氛围下加入溶剂(1.0ml),65℃下搅拌反应48h后,过柱分离制得式(ⅳ)所示的目标产物,详见表3。
[0069]
表3实施例34-91的产物收率、ee值和数据表征
[0070]
[0071]
[0072]
[0073]
[0074]
[0075]
[0076]
[0077]
[0078]
[0079]
[0080]
[0081]
[0082]
[0083]
[0084]
[0085]
[0086]
[0087]
[0088]
[0089][0090]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。