1.本发明涉及植物有效活性成分提取技术领域,具体涉及分子印迹聚合物及其制备方法和应用、从何首乌叶中同时提取三种黄酮苷类化合物的方法。
背景技术:
2.何首乌(polygonum multiflorum thunb.),又名夜交藤,首乌藤,为蓼科何首乌属多年生草本植物何首乌的干燥块茎。何首乌中含有多种生物活性类物质,其中最主要的包括黄酮类化合物、二苯乙烯苷类化合物、蒽醌类化合物、多酚类化合物和磷脂类及脂肪酸类等活性成分。现代药理研究表明,何首乌具有镇静安神、抗慢性炎症及抗菌等作用,其中的黄酮类化合物也有着抗氧化、抗衰老、抗癌和降三高的作用。
3.江苏省食品药品监督检验研究院从何首乌叶中鉴定出6个黄酮主要成分的结构,分别为3-甲氧基-4-羟基苯甲酸-5-o-葡萄糖苷、杨梅苷、槲皮苷、山柰酚-3-o-鼠李糖苷、槲皮素与山柰酚。中国专利cn113402574a公开了高速逆流色谱分离纯化何首乌叶乙酸乙酯相中黄酮类化合物的方法,以水-甲醇-乙酸乙酯-正己烷溶液为溶剂体系,分离得到杨梅苷、槲皮苷和山柰酚-3-o-鼠李糖苷(阿福豆苷),分离纯化黄酮类化合物单体纯度高、损失小、操作简单、经济环保。然而,高速逆流色谱分离纯化过程中,进样浓度为5~20mg/ml,进样体积为10~30ml,杨梅苷、阿福豆苷和槲皮苷三种黄酮类化合物分离量小(0.05~0.6g),制备效率低。
技术实现要素:
4.有鉴于此,本发明的目的在于提供分子印迹聚合物及其制备方法和应用、从何首乌叶中同时提取三种黄酮苷类化合物的方法,本发明提供的分子印迹聚合物对杨梅苷、槲皮苷和阿福豆苷具有高选择性吸附性和高吸附量,以上述分子印迹聚合物对三种黄酮苷类化合物进行分离纯化,分离量大,杨梅苷、阿福豆苷和槲皮苷三种黄酮苷类化合物的制备效率高。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种分子印迹聚合物,包括以下质量份数的制备原料:
7.模板分子1份,功能单体1~50份,金属离子源0.01~0.1份,交联剂0.1~20份,引发剂0.1~3份;
8.所述模板分子为杨梅苷、槲皮苷和阿福豆苷;
9.所述功能单体包括α-甲基丙烯酸、2-乙烯吡啶、4-乙烯基吡啶、n-二乙基胺基甲基丙烯酸乙酯和丙烯胺中的至少一种。
10.优选地,所述金属离子源中的金属离子包括铝离子、铁离子、锌离子、铜离子和钙离子中的至少一种。
11.本发明提供了上述技术方案所述分子印迹聚合物的制备方法,包括以下步骤:
12.将模板分子、功能单体、金属离子和醇水溶液混合,进行配位反应,得到配合物液;
13.将所述配合物液、交联剂和引发剂混合,进行聚合反应后去除模板分子,得到分子印迹聚合物。
14.本发明提供了上述技术方案所述的分子印迹聚合物或上述技术方案所述制备方法制得的分子印迹聚合物在分离纯化黄酮苷类化合物中的应用;所述黄酮苷类化合物包括杨梅苷、槲皮苷和阿福豆苷。
15.本发明提供了一种从何首乌叶中同时提取三种黄酮苷类化合物的方法,包括以下步骤:
16.将何首乌叶进行依次进行醇提、第一脱色除杂、浓缩和分散于水中,得到醇提物水悬浮液;
17.将所述醇提物水悬浮液依次进行脱脂和除弱极性成分,得到水相;
18.将所述水相进行萃取,将得到的有机相进行第二脱色除杂,得到黄酮苷粗品有机相;所述萃取用萃取剂包括乙酸乙酯、乙酸乙酯-甲醇溶剂或乙酸乙酯-乙腈溶剂;
19.将所述黄酮苷粗品有机相与分子印迹聚合物混合,进行吸附,将所述吸附得到黄酮苷-分子印迹聚合物进行洗脱,得到三种黄酮苷类化合物;所述分子印迹聚合物为上述技术方案所述的分子印迹聚合物或上述技术方案所述制备方法制得的分子印迹聚合物;所述三种黄酮苷类化合物为杨梅苷、槲皮苷和阿福豆苷。
20.优选地,所述醇提为将何首乌叶浸泡入乙醇水溶液中,进行间歇闪式提取;
21.所述乙醇水溶液中乙醇的体积分数为50~100%;
22.所述何首乌叶的干重与乙醇水溶液的体积之比为1g:5~20ml;
23.所述醇提的温度为30~60℃,单次闪式提取的时间为10~30min,闪式提取的间隔时间为1~8h。
24.优选地,所述除弱极性成分的方式为二氯甲烷萃取。
25.优选地,所述第一脱色除杂和第二脱色除杂用脱色除杂剂为活性炭;
26.所述第一脱色除杂和第二脱色除杂的温度独立地为50~70℃。
27.优选地,所述黄酮苷粗品有机相的用量以何首乌叶计,何首乌叶与分子印迹聚合物的质量比为1:0.01~0.04;
28.所述吸附的时间为8~36h。
29.优选地,所述洗脱用洗脱剂包括乙腈水溶液、甲醇水溶液。
30.本发明提供了一种分子印迹聚合物(mips),包括以下质量份数的制备原料:模板分子1份,功能单体1~50份,金属离子源0.01~0.1份,交联剂0.1~20份,引发剂0.1~3份;所述模板分子为杨梅苷、槲皮苷和阿福豆苷;所述功能单体包括α-甲基丙烯酸、2-乙烯吡啶、4-乙烯基吡啶、n-二乙基胺基甲基丙烯酸乙酯和丙烯胺中的至少一种。本发明以杨梅苷、槲皮苷和阿福豆苷作为模板分子制备的分子印迹聚合物具有构效预定性;通过引入金属离子,分子印迹聚合物利用金属离子的配位作用,是一种具有较强分子识别能力的新型高分子仿生材料,对杨梅苷、槲皮苷和阿福豆苷具有特异性、高选择识别能力和高吸附能力;在交联剂作用下功能单体与模板分子形成特定的空间结构,且该结构稳定,大大提高了mips的强度。本发明mips提供的具有从复杂样品中选择性提取目标分子或与其结构相近的某一类化合物的能力,适合作为固相萃取填料、固相微萃取涂层以及分子印迹薄膜来分离
富集复杂样品中的痕量分析物,克服样品体系复杂、预处理繁琐等不利因素,达到样品分离纯化的目的,在杨梅苷、槲皮苷和阿福豆苷的制备方面具有很好的应用前景,为杨梅苷、槲皮苷和阿福豆苷的分离纯化提供了一种新途径。
31.本发明提供了上述技术方案所述分子印迹聚合物的制备方法。本发明通过将模板分子与功能单体、交联剂和引发剂在醇水这种特定的分散体系中进行共聚合,制得高交联刚性聚合物,然后通过物理或化学的方法除去其中的模板分子,得到具有确定空间构型的空穴和功能基在空穴内精确排布的聚合物。而且,本发明提供的制备方法,工艺简单,操作简单,制备原料来源广,生产成本低。
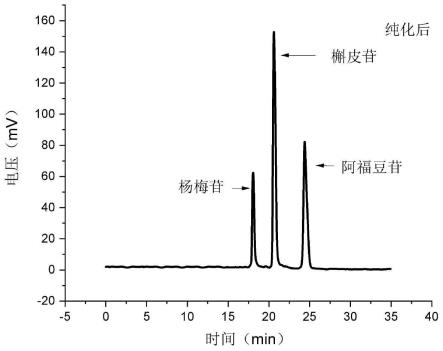
32.本发明提供了一种从何首乌叶中同时提取三种黄酮苷类化合物的方法。分子印迹技术是为获得在空间结构和结合位点上与模板分子完全匹配的聚合物的实验制备技术。本发明利用对杨梅苷、槲皮苷和阿福豆苷具有特异性、高选择识别能力和高吸附能力且结构稳定、强度高的分子印迹聚合物对黄酮苷粗品乙酯相吸附和洗脱即可一次性分离得到杨梅苷、槲皮苷和阿福豆苷三种黄酮苷类化合物,黄酮苷类化合物的分离量大、分离效率高、操作简单;而且分子印迹聚合物重复利用性高,,适合大规模生产。如实施例测试结果所示,本发明提供的方法一次性可制备得到4.43g以上的三种黄酮苷类化合物,目标产物的产量高,制备效率高;三种黄酮苷类化合物的总收率为0.886~0.972%,收率高;三种黄酮苷类化合物的纯度在98.72%以上,纯度高。
附图说明
33.图1为实施例1中黄酮苷粗品乙酯相的液相色谱图;
34.图2为实施例1分离得到的三种黄酮苷类化合物的液相色谱图。
具体实施方式
35.本发明提供了一种分子印迹聚合物,包括以下质量份数的制备原料:模板分子1份,功能单体1~50份,金属离子源0.01~0.1份,交联剂0.1~20份,引发剂0.1~3份。
36.在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
37.以质量份数计,本发明提供的分子印迹聚合物的制备原料包括模板分子1份;所述模板分子为杨梅苷、阿福豆苷和槲皮苷,所述杨梅苷、槲皮苷和阿福豆苷的质量比优选为1:3:1.5~2,更优选为1:3:1.5。
38.以所述模板分子的质量份数计,本发明提供的分子印迹聚合物的制备原料包括功能单体1~50份,优选为10~35份;所述功能单体包括α-甲基丙烯酸、2-乙烯吡啶、4-乙烯基吡啶、n-二乙基胺基甲基丙烯酸乙酯(deam)和丙烯胺中的至少一种。
39.以所述模板分子的质量份数计,本发明提供的分子印迹聚合物的制备原料包括金属离子源0.01~0.1份,优选为0.012~0.07份;所述金属离子源中的金属离子优选包括铝离子、铁离子、锌离子、铜离子和钙离子中的至少一种;本发明对于所述金属离子源没有特殊限定,采用本领域技术人员熟知的的水溶性铝盐、水溶性铁盐、水溶性锌盐、水溶性铜盐和水溶性钙盐即可,具体如硝酸铝、氯化铁、硫酸锌、硫酸铜和氯化钙中的至少一种。如果分子印迹聚合物的制备过程中不加入金属离子,模板分子仅通过非共价结合(氢键、范德华
力),作用力比较弱。而金属离子参与分子印迹聚合物的制备后,形成的配位键比氢键以及范德华力的结合能力更强,以配位作用形成的分子印迹聚合物能依靠中心金属离子的相互作用同时达到热力学和动力学的平衡,因此可以显著提高模板分子的吸附能力。
40.以所述模板分子的质量份数计,本发明提供的分子印迹聚合物的制备原料包括交联剂0.1~20份,优选为6~20份;所述交联剂优选为乙二醇二甲基丙烯酸酯(edma)。
41.以所述模板分子的质量份数计,本发明提供的分子印迹聚合物的制备原料包括引发剂0.1~3份,优选为1~2.5份;所述引发剂优选包括偶氮二异丁腈和/或偶氮二环己基甲腈。
42.在本发明中,所述分子印迹聚合物的粒径优选≤180μm(80目),更优选≤75μm(200目)。
43.本发明提供了上述技术方案所述分子印迹聚合物的制备方法,包括以下步骤:
44.将模板分子、功能单体、金属离子和醇水溶液混合,进行配位反应,得到配合物液;
45.将所述配合物液、交联剂和引发剂混合,进行聚合反应后去除模板分子,得到分子印迹聚合物。
46.本发明将模板分子、功能单体、金属离子和醇水溶液混合(记为第一混合),进行配位反应,得到配合物液。
47.在本发明中,所述醇水溶液优选为甲醇水溶液,所述甲醇水溶液中甲醇的体积分数优选为50~90%,更优选为60~80%;本发明对于所述甲醇水溶液的用量没有特殊限定,使得所述混合得到的模板分子混合液中模板分子的总浓度为0.5~5mmol/l即可,更优选为1~2mmol/l。
48.在本发明中,所述混合优选为超声混合,所述超声混合的温度优选为30~50℃,更优选为40℃,时间优选为5~20min,更优选为10~15min,超声功率优选为500~800w,更优选为600~700w。
49.在本发明中,所述配位反应的温度优选为30~50℃,更优选为40℃;所述配位反应的时间优选为6~24h,更优选为10~15h。
50.得到配合物液后,本发明将所述配合物液、交联剂和引发剂混合(记为第二混合),进行聚合反应后去除模板分子,得到分子印迹聚合物。
51.在本发明中,所述第二混合的方式优选与所述第一混合相同,在此不再赘述。
52.在本发明中,所述聚合反应的温度优选为50~70℃,更优选为60℃,时间优选为24~48h,更优选为24~30h;所述聚合反应优选在保护气氛和密封条件下进行,所述保护气氛优选为氮气、氩气或氦气。
53.完成所述聚合反应后,本发明优选还包括将所得聚合反应液依次进行干燥、研磨和过筛,得到前驱体;所述干燥的温度优选为30~50℃,更优选为40℃,时间优选为6~24h,更优选为10~15h;所述过筛优选为过80~200目的筛。
54.在本发明中,所述去除模板分子优选为利用乙腈-甲醇-水溶液进行回流提取;所述乙腈-甲醇-水溶液中乙腈、甲醇和水的体积比优选为2~3:3~5:3~4;所述回流提取的时间优选为12~24h,更优选为12~15h。
55.去除模板分子后,本发明优选还包括将所得去除模板分子的反应液进行离心分离,将所得固体组分进行超声甲醇洗涤后干燥,得到分子印迹聚合物;所述超声甲醇洗涤的
温度、功率和时间与前述超声混合相同,在此不再赘述。在本发明中,所述干燥的温度优选为30~50℃,更优选为40℃,时间没优选为6~24h,更优选为10~15h。
56.本发明提供了上述技术方案所述分子印迹聚合物或上述技术方案所述制备方法制得的分子印迹聚合物在分离纯化黄酮苷类化合物中的应用;所述黄酮苷类化合物包括杨梅苷、阿福豆苷和槲皮苷。
57.本发明提供了一种从何首乌叶中同时提取三种黄酮苷类化合物的方法,包括以下步骤:
58.将何首乌叶进行依次进行醇提、第一脱色除杂、浓缩和分散于水中,得到醇提物水悬浮液;
59.将所述醇提物水悬浮液依次进行脱脂和除弱极性成分,得到水相;
60.将所述水相进行萃取,将得到的有机相进行第二脱色除杂,得到黄酮苷粗品有机相;所述萃取用萃取剂包括乙酸乙酯、乙酸乙酯-甲醇溶剂或乙酸乙酯-乙腈溶剂;
61.将所述黄酮苷粗品有机相与分子印迹聚合物混合,进行吸附,将所述吸附得到黄酮苷-分子印迹聚合物进行洗脱,得到三种黄酮苷类化合物;所述分子印迹聚合物为上述技术方案所述的分子印迹聚合物或上述技术方案所述制备方法制得的分子印迹聚合物;所述三种黄酮苷类化合物为杨梅苷、槲皮苷和阿福豆苷。
62.本发明将何首乌叶进行依次进行醇提、第一脱色除杂、浓缩和分散于水中,得到醇提物水悬浮液。
63.在本发明中,所述醇提优选为将何首乌叶浸泡入乙醇水溶液中,进行间歇闪式提取,所述乙醇水溶液中乙醇的体积分数优选为50~100%,更优选为60~90%;所述何首乌叶的干重与乙醇水溶液的体积之比优选为1g:5~20ml,更优选为1g:8~15ml;所述醇提的温度优选为30~60℃,更优选为40~50℃,所述醇提优选在搅拌条件下进行,所述搅拌的速度优选为2000~4000rpm,更优选为3000rpm;所述浸泡的时间优选为12~72h,更优选为20~60h,进一步优选为40~48h;单次闪式提取的时间为10~30min,更优选为10~20min,进一步优选为10~15min,所述闪式提取的间隔时间为1~8h,更优选为2~7h,进一步优选为4~6h;本发明采用闪式提取方式能够将物料打碎,提高黄酮苷类化合物的收率。
64.在本发明中,所述第一脱色除杂用脱色除杂剂优选为活性炭,所述何首乌叶与活性炭的质量比优选为1:0.1~0.3,更优选为1:0.1~0.15;所述第一脱色除杂的温度优选为50~70℃,更优选为60℃,时间优选为1~4h,更优选为2~3h;所述第一脱色除杂优选在搅拌条件下进行。
65.完成所述第一脱色除杂后,本发明优选还包括固液分离,将所得液体组分再进行浓缩。本发明进行第一脱色除杂能够去除部分杂质,利于后续的固液分离,进而有利于提高三种黄酮苷类化合物的收率和纯度。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如离心分离。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压浓缩。
66.本发明对于所述水的用量没有特殊限定,能够将所述浓缩得到的粗浸膏分散均匀即可。
67.得到醇提物水悬浮液后,本发明将所述醇提物水悬浮液依次进行脱脂和除弱极性成分,得到水相。
68.在本发明中,所述脱脂用脱脂剂优选包括石油醚和/或正己烷,所述醇提物水悬浮液与脱脂剂的体积比优选为1:0.5~2,更优选为1:1~1.5。
69.在本发明中,所述除弱极性成分的方式优选为二氯甲烷萃取;本发明对于所述二氯甲烷萃取的次数没有特殊限定,以最后一次二氯甲烷萃取得到的二氯甲烷相无色为准,所述醇提物水悬浮液与单次二氯甲烷萃取用二氯甲烷的体积比优选为1:0.5~2,更优选为1:1~1.5。
70.得到水相后,本发明将所述水相进行萃取,将得到的有机相进行第二脱色除杂,得到黄酮苷粗品有机相;所述萃取用萃取剂包括乙酸乙酯、乙酸乙酯-甲醇溶剂或乙酸乙酯-乙腈溶剂。
71.在本发明中,本发明对于所述萃取的次数没有特殊限定,以最后一次萃取时有机相无色为准,所述水相与单次萃取用萃取剂的体积比优选为1:0.5~2,更优选为1:1~1.5。在本发明中,所述乙酸乙酯-甲醇溶剂中乙酸乙酯与甲醇的体积比优选为1:0.01~0.1,更优选为1:0.05;所述乙酸乙酯-乙腈溶剂中乙酸乙酯与乙腈的体积比优选为1:0.01~0.1,更优选为1:0.05。
72.在本发明中,所述第二脱色除杂的条件优选与所述第一脱色除杂条件相同,在此不再赘述。完成所述第二脱色除杂后,本发明优选还包括固液分离,所得液体组分为黄酮苷粗品有机相。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如离心分离。
73.得到黄酮苷粗品乙酯相后,本发明将所述黄酮苷粗品有机相与分子印迹聚合物混合,进行吸附,将所述吸附得到黄酮苷-分子印迹聚合物进行洗脱,得到三种黄酮苷类化合物;所述分子印迹聚合物为上述技术方案所述的分子印迹聚合物或上述技术方案所述制备方法制得的分子印迹聚合物;所述三种黄酮苷类化合物为杨梅苷、阿福豆苷和槲皮苷。
74.在本发明中,所述黄酮苷粗品有机相的用量以何首乌叶计,何首乌叶与分子印迹聚合物的质量比优选为1:0.01~0.04,更优选为1:0.03~0.04。
75.在本发明中,所述吸附的温度优选为室温,所述吸附的时间优选为8~36h,更优选为10~30h,进一步优选为24h;所述吸附优选在振荡条件下进行。
76.所述吸附后本发明优选还包括将所得吸附体系进行固液分离,所得固体组分为黄酮苷-分子印迹聚合物。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如离心分离。
77.在本发明中,所述洗脱用洗脱剂优选包括乙腈水溶液和/或甲醇水溶液;所述乙腈水溶液中乙腈的体积分数优选为10~50%,更优选为20~40%;所述甲醇水溶液中甲醇的体积分数优选为10~50%,更优选为20~40%。
78.所述洗脱后,本发明优选还包括将所得洗脱液浓缩至恒重,得到三种黄酮苷类化合物。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压浓缩。
79.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
80.实施例1
81.(1)将0.5g模板分子(杨梅苷、槲皮苷、阿福豆苷质量比=1:3:1.5)、5g deam、5g 2-乙烯吡啶、0.01g硝酸铝和70%(v/v)甲醇水溶液混合均匀,得到模板分子总浓度为0.1mmol/l的模板分子混合液,在40℃、600w条件下超声处理15min,静置12h,然后加入10g edma和1g偶氮二异丁腈,继续超声处理15min,通入氮气15min后密封,在60℃水浴条件下反应24h,真空干燥,研磨,过200目分子筛,筛下部分用乙腈:甲醇:水体积比=3:3:4的混合溶剂回流提取12h,离心分离,所得固体组分在40℃、600w超声甲醇洗涤15min,在40℃条件下干燥12h,得到分子印迹聚合物。
82.(2)将500g何首乌叶枝粉置于60%(v/v)的乙醇水溶液(料液比为1:10(g/ml)),在40℃条件下浸泡48h,浸泡期间进行闪式提取,单次提取时间为10min,搅拌速度为3000rpm,闪式提取间隔时间为6h,加入50g活性炭(记为第一活性炭),在60℃、搅拌条件下第一脱色除杂2h,离心分离,将所得液体组分进行减压浓缩,将所得粗浸膏分散于水中,将所得醇提物水悬浮液进行石油醚脱脂(醇提物水悬浮液与石油醚体积比为1:1),然后进行二氯甲烷萃取(醇提物水悬浮液与二氯甲烷体积比为1:1)至最后一次萃取所得二氯甲烷相无色,将所得水相进行乙酸乙酯萃取(醇提物水悬浮液与乙酸乙酯体积比为1:1)至最后一次萃取所得有机相无色,在所得乙酯相中加入20g活性炭(记为第二活性炭),在60℃、搅拌条件下第二脱色除杂2h,离心分离,在所得黄酮苷粗品乙酯相中加入所述分子印迹聚合物,在室温条件下下振荡24h,离心分离,所得固体组分为黄酮苷-分子印迹聚合物;利用30%(v/v)乙腈水溶液对所述黄酮苷-分子印迹聚合物进行洗脱,将所得洗脱液浓缩至恒重,得到三种黄酮苷类化合物(杨梅苷、阿福豆苷和槲皮苷)。
83.图1为黄酮苷粗品乙酯相的液相色谱图,图2为分离得到的三种黄酮苷类化合物的色谱液相图,由图1~2可知,本发明提供的方法能够一次性提取得到三种黄酮苷类化合物(杨梅苷、阿福豆苷和槲皮苷)。
84.实施例2~4
85.按照实施例1的方法提取三种黄酮苷类化合物,实施例2~4的制备条件如表1所示,其他提取条件与实施例1相同。
86.对比例1
87.按照实施例4的方法提取三种黄酮苷类化合物,对比例1制备条件如表1所示,其他提取条件与实施例4相同。
88.表2实施例1~5的提取条件以及三种黄酮苷类化合物的提取效果
[0089][0090]
由表1中实施例2~4可知,本发明提供的方法一次性可制备得到4.4g以上的三种黄酮苷类化合物,三种黄酮苷类化合物的总收率为0.886~0.972%,纯度在98.72%以上,说明,本发明提供的方法三种黄酮苷类化合物的产量高,提取效率高,收率高,纯度高。而不加金属离子源的分子印迹聚合物提取黄酮苷的质量远小于含有金属离子源的分子印迹聚合物对黄酮苷类化合物的提取效率。
[0091]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。