用于高效表达纯化小标签活性融合蛋白的gateway原核载体系统
技术领域
1.本发明涉及一件快速高效表达活性蛋白的gateway原核载体系统。
背景技术:
2.gateway系统是一套高效精确的分子克隆技术。该套系统只需要两个简单的lr或bp反应,就能使目标序列在表达载体(destination vector)和入门载体(entr vector)之间随意高效准确转换,且能保证每个目标序列的旁侧序列都高度一致。蛋白质的活性功能是生命力的体现。研究蛋白的结构,功能和活性一直是生物学研究的基本目标之一。将某一功能蛋白在非变性条件下纯化,测试其各种生物属性,是实现这一基本目标的主要途径。但现有的蛋白原核表达载体系统,往往采用低效过时的酶切连接系统,耗时费力,筛选克隆时假阳性比例高,而且不能保证每种表达出来的小标签融合蛋白都有完全一致的旁侧连接序列,对照性不强。因此,我们希望开发一套能利用gateway分子克隆技术的新的载体系统,帮助我们更加高效精确的表达大量活性目标蛋白,杜绝分子克隆过程中的假阳性问题,并解决不同融合蛋白之间的旁侧连接序列不一致的问题,提高可对照性,为纯化活性功能蛋白做好前期工作。
技术实现要素:
3.本发明的目的在于提供一种用于高效表达及纯化小标签活性融合蛋白的gateway原核载体系统,以解决上述背景技术中提出的问题。另外,此发明为高效的原核表达系统,待专利批准后,将与本研究组另行发明的真核表达系统(专利申请号202110577823.0)合并开发成可商业化的试剂盒套装。此试剂盒套装可用于所有植物源活性蛋白的表达纯化,以及绝大多数动物源/人源活性蛋白的表达纯化。
4.为解决上述技术问题,本发明开发出了可与入门载体(entr vector)配合使用的空目标表达载体(destination vector):6xhis-gw
s-6xhis,用于高效表达纯化小标签活性融合蛋白的gateway原核载体系统,其具体开发方案如下:
5.1)以pet30a载体为底物,利用限制性内切酶bamhi和hindiii对其进行双酶切,得到酶切产物,并对酶切后的主片段进行纯化;
6.2)设计带有重组位点的gateway座位序列扩增正向引物pet30a-gwf(5
’‑
ccatggctgatatcggatccacaagtttgtacaaaaaagc-3’)以及反向引物pet30a-gwr(5
’‑
gtgcggccgcaagcttgaccactttgtacaagaaagctg-3’),以载体pb2gw7为模板,扩增带有attr1和attr2两个lr反应位点,以及cmr和ccdb两个开放阅读框的pcr片段;
7.3)检测pcr产物,并将正确片段长度的pcr产物纯化;
8.4)将纯化后的pet30a主片段和纯化后的pcr产物按一定比例混合,加入重组酶multis和buffer,配制成反应体系,37度恒温反应半小时;
9.5)将反应好的重组产物导入大肠杆菌db3.1感受态细胞,加入适量soc液态培养
基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有卡那霉素的固体lb培养基平板上,并放入37度生长箱过夜培养;
10.6)设计一条位于pet30a骨架上t7启动子内的正向引物t7f(5
’‑
ttaatacgactcactatag-[0011]3’
),以及一条位于pcr连接产物上的反向引物cmrr1(5
’‑
atcacagacggcatgatgaa-3’),用于对单克隆的pcr检测;
[0012]
7)培养皿平板上长出适当大小的克隆后,挑取部分单克隆溶于20ul水中,取其中1.5ul作为样品模板,利用设计好的一对检测引物,配制成20ul pcr反应体系,对这些克隆进行检测,若pcr能扩增出预期大小的片段,则说明重组反应可能成功;
[0013]
8)挑取能扩增出预期大小片段的单克隆接种于5ml含卡那霉素的lb液体培养基中,37度摇床200rpm/min过夜培养;
[0014]
9)利用试剂盒提取单克隆质粒;
[0015]
10)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明gateway系统元件及ccdb和cmr基因添加成功,得6xhis-gw
k-6xhis载体;
[0016]
(利用此6xhis-gw
k-6xhis载体,与包含目标基因的非卡那霉素抗性的entr载体进行lr反应,利用含卡那霉素的lb培养基平板筛选,即可获得最终表达载体。但若包含目标基因片段的entr载体本身已具有卡那霉素抗性基因,就不能再利用6xhis-gw
k-6xhis通过lr反应构建最终表达载体,而需用到我们最终开发出的具有奇霉素(spectinmycin)抗性的空目标表达载体6xhis-gw
s-6xhis。)
[0017]
在6xhis-gw
k-6xhis的制作基础上,6xhis-gw
s-6xhis载体经历了以下更多步骤,从而最终获得:
[0018]
11)以6xhis-gw
k-6xhis载体为底物,利用限制性内切酶fspi对其进行酶切,得到酶切产物,并对酶切后片段进行纯化;
[0019]
12)设计合成带有重组位点的正向引物specrf3(5
’‑
[0020]
gacaggagcacgatcatgcgcaggcacgaacccagtggacata-3’)以及反向引物specrr3(5
’‑
atggcggccccacgggtgcgcagtcatgcatgatatatctcccaa-3’),以载体pb2gw7为模板,扩增带有启动子,终止子以及开放阅读框的整个奇霉素抗性基因pcr片段;
[0021]
13)检测pcr产物,并将正确片段长度的pcr产物纯化;
[0022]
14)将纯化后的6xhis-gw
k-6xhis酶切片段和纯化后的pcr产物按一定比例混合,加入重组酶multis和buffer,配制成反应体系,37度恒温反应半小时;
[0023]
15)将反应好的重组产物导入大肠杆菌db3.1感受态细胞,加入适量soc液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有奇霉素的固体lb培养基平板上,并放入37度生长箱过夜培养;
[0024]
16)挑选正常生长的单克隆接种于5ml含奇霉素的lb液体培养基中,37度摇床200rpm/min过夜培养;
[0025]
17)利用试剂盒提取单克隆质粒;
[0026]
18)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明产品开发成功,得6xhis-gw
s-6xhis,大量留存该质粒,等待分子克隆制作某一特定蛋白表达载体时使用。(通过将6xhis-gw
s-6xhis载体与包含目标基因的非奇霉素抗性的entr载
体进行lr反应,利用含奇霉素的lb培养基平板筛选,可获得最终表达载体。)
[0027]
本发明优点在于:本发明可用于各种情况下单一蛋白成分的大量原核表达。蛋白两端都有分子量很小,不影响蛋白功能的组蛋白标签(6xhis),更利于蛋白高效过柱纯化;通过加入ccdb基因及cmr基因,杜绝了分子克隆过程中的假阳性问题;通过加入gateway系统的attr1及attr2序列位点,解决了不同融合蛋白之间旁侧连接序列不一致的问题,提高了可对照性,具有高效准确的特点;通过加入奇霉素抗性基因,使得6xhis-gw
s-6xhis载体内,有了除使用卡那霉素抗性基因的第二种选择,让我们能自由地选择几乎所有的商业化entr载体进行lr反应。总之,此发明能极大地推动促进活性蛋白的纯化工作,为蛋白的功能学研究建立基础。
附图说明
[0028]
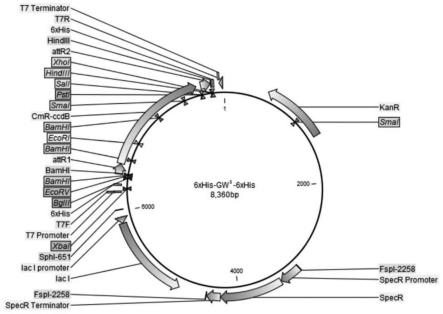
图1是6xhis-gw
s-6xhis载体的结构示意图。
[0029]
图2是6xhis-gw
k-6xhis载体制作流程示意图。
[0030]
图3是在6xhis-gw
k-6xhis基础上制作6xhis-gw
s-6xhis载体的流程示意图。
具体实施方式
[0031]
下面用具体实施例说明本发明,并不是对本发明的限制。
[0032]
实施例
[0033]
一件快速高效表达活性蛋白的gateway系统原核表达载体——6xhis-gw
s-6xhis。具体制作方法包括以下内容:
[0034]
1)以pet30a载体为底物,利用限制性内切酶bamhi和hindiii对其进行双酶切,得到酶切产物,并对酶切后的主片段进行纯化;
[0035]
2)设计带有重组位点的gateway座位序列扩增正向引物pet30a-gwf(5
’‑
ccatggctgatatcggatccacaagtttgtacaaaaaagc-3’)以及反向引物pet30a-gwr(5
’‑
gtgcggccgcaagcttgaccactttgtacaagaaagctg-3’),以载体pb2gw7为模板,扩增带有attr1和attr2两个lr反应位点,以及cmr和ccdb两个开放阅读框的pcr片段;
[0036]
3)检测pcr产物,并将正确片段长度的pcr产物纯化;
[0037]
4)将纯化后的pet30a主片段和纯化后的pcr产物按一定比例混合,加入重组酶multis和buffer,配制成反应体系,37度恒温反应半小时;
[0038]
5)将反应好的重组产物导入大肠杆菌db3.1感受态细胞,加入适量soc液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有卡那霉素的固体lb培养基平板上,并放入37度生长箱过夜培养;
[0039]
6)设计一条位于pet30a骨架上t7启动子内的正向引物t7f(5
’‑
ttaatacgactcactatag-[0040]3’
),以及一条位于pcr连接产物上的反向引物cmrr1(5
’‑
atcacagacggcatgatgaa-3’),用于对单克隆的pcr检测;
[0041]
7)培养皿平板上长出适当大小的克隆后,挑取部分单克隆溶于20ul水中,取其中1.5ul作为样品模板,利用设计好的一对检测引物,配制成20ul pcr反应体系,对这些克隆进行检测,若pcr能扩增出预期大小的片段,则说明重组反应可能成功;
[0042]
8)挑取能扩增出预期大小片段的单克隆接种于5ml含卡那霉素的lb液体培养基中,37度摇床200rpm/min过夜培养;
[0043]
9)利用试剂盒提取单克隆质粒;
[0044]
10)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明gateway系统元件及ccdb和cmr基因添加成功,得6xhis-gw
k-6xhis载体;
[0045]
(利用此6xhis-gw
k-6xhis载体,与包含目标基因的非卡那霉素抗性的entr载体进行lr反应,利用含卡那霉素的lb培养基平板筛选,即可获得最终表达载体。但若包含目标基因片段的entr载体本身已具有卡那霉素抗性基因,就不能再利用6xhis-gw
k-6xhis通过lr反应构建最终表达载体,而需用到我们最终开发出的具有奇霉素(spectinmycin)抗性的空目标表达载体6xhis-gw
s-6xhis。)
[0046]
在6xhis-gw
k-6xhis的制作基础上,6xhis-gw
s-6xhis载体经历了以下更多步骤,从而最终获得:
[0047]
11)以6xhis-gw
k-6xhis载体为底物,利用限制性内切酶fspi对其进行酶切,得到酶切产物,并对酶切后片段进行纯化;
[0048]
12)设计合成带有重组位点的正向引物specrf3(5
’‑
[0049]
gacaggagcacgatcatgcgcaggcacgaacccagtggacata-3’)以及反向引物specrr3(5
’‑
atggcggccccacgggtgcgcagtcatgcatgatatatctcccaa-3’),以载体pb2gw7为模板,扩增带有启动子,终止子以及开放阅读框的整个奇霉素抗性基因pcr片段;
[0050]
13)检测pcr产物,并将正确片段长度的pcr产物纯化;
[0051]
14)将纯化后的6xhis-gw
k-6xhis酶切片段和纯化后的pcr产物按一定比例混合,加入重组酶multis和buffer,配制成反应体系,37度恒温反应半小时;
[0052]
15)将反应好的重组产物导入大肠杆菌db3.1感受态细胞,加入适量soc液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有奇霉素的固体lb培养基平板上,并放入37度生长箱过夜培养;
[0053]
16)挑选正常生长的单克隆接种于5ml含奇霉素的lb液体培养基中,37度摇床200rpm/min过夜培养;
[0054]
17)利用试剂盒提取单克隆质粒;
[0055]
18)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明产品开发成功,得6xhis-gw
s-6xhis,大量留存该质粒,等待分子克隆制作某一特定蛋白表达载体时使用。
[0056]
(通过将6xhis-gw
s-6xhis载体与包含目标基因的非奇霉素抗性的entr载体进行lr反应,利用含奇霉素的lb培养基平板筛选,可获得最终表达载体。)
[0057]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。