1.本公开的实施例总体涉及生物信息学领域,并且更具体地涉及一种用于构建微生物基因组数据库的方法、设备和介质。
背景技术:
2.目前,在需要进行微生物物种鉴定时,通常是基于公共数据库来进行比对的。然而,由于一般的公共数据库均仅包括有限的微生物物种,而且存在数据量大并且数据质量参差不齐的问题,因此微生物物种鉴定的比对率一般,准确性较低,而且比对效率也非常低。
3.另外,现有的私有微生物基因组数据库通常都是基于筛选出的高质量的基因组来构建的,因此存在物种缺失的问题,从而使得在基于这样的私有微生物基因组数据库进行微生物物种鉴定时,容易造成假阴性的问题,因此微生物物种鉴定的比对率也较低。
技术实现要素:
4.针对上述问题,本公开提供了一种用于构建微生物基因组数据库的方法、设备和介质,使得所构建的微生物基因组数据库能够同时具有较高的比对率和比对效率,也就是说所构建的微生物基因组数据库质量高、数据量小、并且数据覆盖范围全面,进而有助于提高微生物物种鉴定的准确性,并且提升比对性能。
5.根据本公开的第一方面,提供了一种用于构建微生物基因组数据库的方法,包括:获取来自多个公共数据库的关于具有文献支持的微生物物种的基因组数据,所述基因组数据包括每一基因组所属的微生物物种、所述微生物物种所属的微生物类别、所述基因组的组装水平、所述基因组的序列数据;基于所获取的基因组数据所涉及的多个微生物类别,对所获取的基因组数据进行初步筛选;基于每一基因组的组装水平,将经初步筛选的基因组数据分组成与第一组装水平相关联的第一基因组数据分组以及分别与多个第二组装水平相关联的多个第二基因组数据分组,所述第一组装水平的级别高于所述多个第二组装水平;从所述多个第二基因组数据分组中的至少一个中标识出病原微生物物种的基因组数据,以便基于所标识出的基因组数据以及所述第一基因组数据分组确定经最终筛选的基因组数据;以及对经最终筛选的基因组数据进行去冗余处理,以便得到用于构建微生物基因组数据库的基因组数据。
6.根据本公开的第二方面,提供了一种计算设备,包括:至少一个处理器;以及与所述至少一个处理器通信连接的存储器;所述存储器存储有可被所述至少一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器能够执行本公开的第一方面的方法。
7.在本公开的第三方面中,提供了一种存储有计算机指令的非瞬时计算机可读存储介质,其中所述计算机指令用于使计算机执行本公开的第一方面的方法。
8.在一些实施例中,对经筛选的基因组数据进行去冗余处理包括:对于任一微生物
物种,如果在经最终筛选的基因组数据中包括与所述微生物物种的两个或更多个基因组相关联的基因组数据,则从所述两个或更多个基因组中确定所述微生物物种的基础基因组;基于所述两个或更多个基因组中除所述基础基因组以外的每一基因组的片段序列,根据预定长度和预定步长将相应基因组打断成多个片段序列,其中每一片段序列的长度等于所述预定长度;将分割得到的每一片段序列与所述基础基因组进行比对,以便确定所述片段序列是否为与所述基础基因组的相似度小于预定阈值的特异性序列;基于为所述微生物物种确定的特异性序列以及所述基础基因组,确定所述微生物物种的完整基因组;以及将与所述完整基因组相关联的基因组数据保存到所述微生物基因组数据库中,作为所述微生物物种的基因组数据。
9.在一些实施例中,基于为所述微生物物种确定的特异性序列以及所述基础基因组,确定所述微生物物种的完整基因组包括:将为所述微生物物种确定的所有特异性序列中具有连续性特征的多个特异性序列进行组装,以便得到多个经组装的特异性序列;将所述多个经组装的特异性序列根据长度进行排序,以便将所述多个经组装的特异性序列中排名靠后的一个或多个经组装的特异性序列丢弃,其中被丢弃的多个经组装的特异性序列的长度之和小于或等于所有特异性序列的总长度的预定比例;以任意顺序将剩下的经组装的特异性序列和所述基础基因组整合成所述微生物物种的完整基因组。
10.在一些实施例中,以任意顺序将剩余的经组装的特异性序列和所述基础基因组整合成所述微生物物种的完整基因组包括:通过将剩余的经组装的特异性序列全部置于所述基础基因组的最前面或者全部至于所述基础基因组的最后面来得到所述完整基因组。
11.在一些实施例中,从所述两个或更多个基因组中确定所述微生物物种的基础基因组包括:确定所述两个或更多个基因组中的每一基因组的组装水平;从所述两个或更多个基因组中标识出组装水平的级别最高的一个或多个基因组;确定所标识的一个或多个基因组的文献支持数量;将所标识的一个或多个基因组中具有最大文献支持数量的基因组确定为所述微生物物种的基础基因组。
12.在一些实施例中,所述预定长度大于等于10bp并且小于等于300bp,并且所述预定步长大于等于1bp。
13.在一些实施例中,在对经筛选的基因组数据进行去冗余处理之前,还包括从所述经筛选的基因组数据中去除包括与质粒序列相关联的基因组数据。
14.在一些实施例中,基于所获取的基因组数据所涉及的多个微生物类别,对所获取的基因组数据进行初步筛选包括:从所获取的基因组数据所涉及的所述多个微生物类别中标识出在物种分类系统中被错误分类的一个或多个微生物类别;以及从所获取的基因组数据中移除与所标识出的一个或多个微生物类别相关联的基因组数据,以便得到所述经初步筛选的基因组数据。
15.在一些实施例中,从所述多个第二基因组数据分组中的至少一个中标识出病原微生物物种的基因组数据包括:从所述多个第二基因组数据分组中与级别相对最高的组装水平相关联的一个第二基因组数据分组中标识出病原微生物物种的基因组数据。
16.在一些实施例中,具有文献支持的微生物物种指示所述微生物物种在大于或等于阈值数量的文献中具有相应的记录。
17.应当理解,本部分所描述的内容并非旨在标识本公开的实施例的关键或重要特
征,也不用于限制本公开的范围。本公开的其它特征将通过以下的说明书而变得容易理解。
附图说明
18.结合附图并参考以下详细说明,本公开各实施例的上述和其他特征、优点及方面将变得更加明显。在附图中,相同或相似的附图标注表示相同或相似的元素。
19.图1示出了用于实现根据本发明的实施例的用于构建微生物基因组数据库的方法的示例性系统100的示意图。
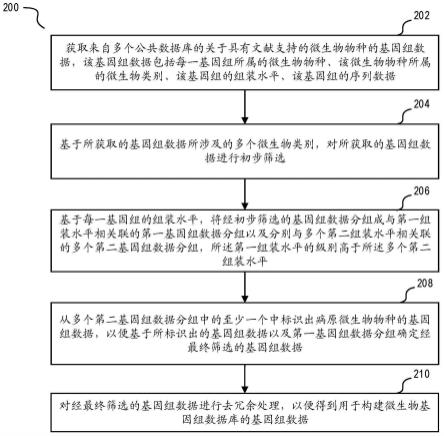
20.图2示出了根据本公开的实施例的用于构建微生物基因组数据库的方法200的流程图。
21.图3示出了根据本公开的实施例的用于对所获取的基因组数据进行初步筛选的方法300的流程图。
22.图4示出了根据本公开的实施例的用于对经筛选的基因组数据进行去冗余处理的方法400的流程图。
23.图5示出了根据本公开的实施例的用于从两个或更多个基因组中确定微生物物种的基础基因组的方法500的流程图。
24.图6示出了根据本公开的实施例的电子设备600的框图。
具体实施方式
25.以下结合附图对本公开的示范性实施例做出说明,其中包括本公开实施例的各种细节以助于理解,应当将它们认为仅仅是示范性的。因此,本领域普通技术人员应当认识到,可以对这里描述的实施例做出各种改变和修改,而不会背离本公开的范围和精神。同样,为了清楚和简明,以下的描述中省略了对公知功能和结构的描述。
26.在本文中使用的术语“包括”及其变形表示开放性包括,即“包括但不限于”。除非特别申明,术语“或”表示“和/或”。术语“基于”表示“至少部分地基于”。术语“一个示例实施例”和“一个实施例”表示“至少一个示例实施例”。术语“另一实施例”表示“至少一个另外的实施例”。术语“第一”、“第二”等等可以指代不同的或相同的对象。下文还可能包括其他明确的和隐含的定义。
27.如上所述,在需要进行微生物物种鉴定时,通常是基于公共数据库来进行比对的。然而,由于一般的公共数据库均仅包括有限的微生物物种,而且存在数据量大并且数据质量参差不齐的问题,因此微生物物种鉴定的比对率一般,而且比对效率也非常低,而且比对效率也非常低。另外,现有的私有微生物基因组数据库通常都是基于筛选出的高质量的基因组来构建的,因此存在物种缺失的问题,从而使得在基于这样的私有微生物基因组数据库进行微生物物种鉴定时,容易造成假阴性的问题,因此微生物物种鉴定的比对率也较低。
28.为了至少部分地解决上述问题以及其他潜在问题中的一个或者多个,本公开的示例实施例提出了一种用于构建微生物基因组数据库的方法,包括:获取来自多个公共数据库的关于具有文献支持的微生物物种的基因组数据,所述基因组数据包括每一基因组所属的微生物物种、所述微生物物种所属的微生物类别、所述基因组的组装水平、所述基因组的序列数据;基于所获取的基因组数据所涉及的多个微生物类别,对所获取的基因组数据进行初步筛选;基于每一基因组的组装水平,将经初步筛选的基因组数据分组成与第一组装
水平相关联的第一基因组数据分组以及分别与多个第二组装水平相关联的多个第二基因组数据分组,所述第一组装水平的级别高于所述多个第二组装水平;从所述多个第二基因组数据分组中的至少一个中标识出病原微生物物种的基因组数据,以便基于所标识出的基因组数据以及所述第一基因组数据分组确定经最终筛选的基因组数据;以及对经最终筛选的基因组数据进行去冗余处理,以便得到用于构建微生物基因组数据库的基因组数据。以此方式,使得所构建的微生物基因组数据库能够同时具有较高的比对率和比对效率,也就是说所构建的微生物基因组数据库质量高、数据量小、并且数据覆盖范围全面,进而有助于提高微生物物种鉴定的准确性,并且提升比对性能。
29.图1示出了用于实现根据本发明的实施例的用于构建微生物基因组数据库的方法的示例性系统100的示意图。如图1中所示,系统100包括计算设备110、网络120以及一个或多个服务器130。计算设备110和该一个或多个服务器130可以例如通过网络120(例如,因特网)进行数据交互。在本公开中,一个或多个服务器130可用于向计算设备110提供从多个公共数据库获取的微生物物种的基因组数据。在本公开中,公共数据库可以是诸如参考序列数据库(ncbi reference sequence database,此后简称为refseq)或者基因银行(genbank)之类的用于记录各种生物(包括微生物)的基因序列数据的各种公共数据库。计算设备110可经由网络120与服务器130进行通信,以从服务器130处获取微生物物种的基因组数据。计算设备110可以包括至少一个处理器112以及与该至少一个处理器112耦合的至少一个存储器114,该存储器114中存储有可由该至少一个处理器112执行的指令116,该指令116在被该至少一个处理器112执行时执行如下所述的方法200。计算设备110的具体结构例如可以如下结合图6所述。当然,在一些实施例中,计算设备110也可以是服务器130的一部分。在另一些实施例中,计算设备110也可以直接从多个公共数据库获取微生物物种的基因组数据。在本公开中,由于计算设备110获取的基因组数据是从多个数据库获取的,因此有助于弥补公共数据库微生物物种有限的缺点。
30.图2示出了根据本公开的实施例的用于构建微生物基因组数据库的方法200的流程图。方法200可由如图1所示的计算设备110执行,也可以在图6所示的电子设备600处执行。应当理解的是,方法200还可以包括未示出的附加框和/或可以省略所示出的框,本公开的范围在此方面不受限制。
31.在步骤202,获取来自多个公共数据库的关于具有文献支持的微生物物种的基因组数据。在本公开中,所获取的基因组数据可包括每一基因组所属的微生物物种的名称、该微生物物种所属的微生物类别、该基因组的组装水平、该基因组的序列数据等等。
32.在本公开中,具有文献支持的微生物物种可指示该微生物物种在大于或等于阈值数量(例如,3个或更多个)的文献中具有相应的记录。在一些实施例中,可先从多个公共数据库中获取关于微生物物种的基因组数据,然后再利用爬虫技术在生物医学文献数据库中对与所获取的基因组数据有关的微生物物种进行批量检索以确定其中具有文献支持的微生物物种,以便在所获取的基因组数据中选出具有文献支持的微生物物种的基因组数据。在另一些实施例中,也可先例如利用爬虫技术在生物医学文献数据库中检索具有文献支持的微生物物种,然后再从多个公共数据库中获取关于这些具有文献支持的微生物物种的基因组数据。
33.应了解,取代基因组的序列数据,基因组数据也可提供基因组的序列数据的获取
地址,以使得基于该基因组数据可有效获得相应的序列数据。
34.如前所述,在本公开中,公共数据库可以是诸如参考序列数据库(ncbi reference sequence database,此后简称为refseq)或者基因银行(genbank)之类的用于记录各种生物(包括微生物)的基因序列数据的各种公共数据库。
35.微生物通常包含:细菌、放线菌、蓝细菌、衣原体、支原体、立克次氏体、螺旋体、真菌、病毒、原生动物等类别。因此,微生物物种可以是诸如新型隐球菌、烟曲霉、白色念珠菌之类的各种真菌,诸如沙门氏菌之类的各种细菌,诸如乙肝病毒之类的各种病毒等等。因此,在本公开中,可从公共数据库获取以上各种微生物物种的基因组数据。
36.物种类别的划分通常是按界门纲目科属种来进行划分的,因此在本公开中微生物类别指的是该微生物物种所属的界、门、纲、目、科、属和/或种等。
37.基因组的组装水平可包括完整基因组(complete genome)水平、染色体水平(即chromosome水平)、scaffold水平和contig水平等多种级别。
38.在步骤204,基于所获取的基因组数据所涉及的多个微生物类别,对所获取的基因组数据进行初步筛选。
39.在本公开中,通过基于微生物类别对所获取的基因组数据进行初步筛选,可以从所获取的基因组数据中移除与被错误分类(即分类不准确)的微生物物种相关联的基因组数据,这有助于提高待构建的微生物基因组数据库的准确性。
40.下面将结合图3对步骤204作进一步更详细的描述。
41.在步骤206,基于每一基因组的组装水平,将经初步筛选的基因组数据分组成与第一组装水平相关联的第一基因组数据分组以及分别与多个第二组装水平相关联的多个第二基因组数据分组。在本公开中,该第一组装水平的级别高于该多个第二组装水平。
42.例如,在本公开中,第一组装水平为组装级别最高的“完整基因组”,所有其他组装水平(例如,chromosome水平、scaffold水平和contig水平等)都被称为第二组装水平。
43.通过步骤206,可从经初步筛选的基因组数据中筛选出高质量基因组,以便保证待构建的微生物基因组数据库主要是基于与高质量基因组相关联的基因组数据来构建的,从而有助于进一步提高该数据库的准确性,减少假阳性的检索结果。
44.在步骤208,从该多个第二基因组数据分组中的至少一个中标识出病原微生物物种的基因组数据,以便基于所标识出的基因组数据以及该第一基因组数据分组确定经最终筛选的基因组数据。
45.在一些实施例中,从该多个第二基因组数据分组中的至少一个中标识出病原微生物物种的基因组数据包括从该多个第二基因组数据分组中与级别相对最高的组装水平相关联的一个第二基因组数据分组中标识出病原微生物物种的基因组数据。由于在步骤202获取的基因组数据是关于具有文献支持的微生物物种的基因组数据,因此这里从该多个第二基因组数据分组中的至少一个中标识出的病原微生物物种的基因组数据必定指的是具有文献支持的病原微生物物种的基因组数据。
46.当然,在本公开中,也可从该多个第二基因组数据分组中与级别相对最高的两个或更多个组装水平相关联的第二基因组数据分组中标识出病原微生物物种的基因组数据。同样,这里从该多个第二基因组数据分组中与级别相对最高的两个或更多个组装水平相关联的第二基因组数据分组中标识出的病原微生物物种的基因组数据必定指的也是具有文
献支持的病原微生物物种的基因组数据。
47.应了解,在构建微生物基因组数据库时,如果仅考虑高质量基因组,容易造成所构建的微生物基因组数据库物种缺失,尤其使得某些病原体微生物的基因组数据无法被所构建的微生物基因组数据库覆盖到,进而造成后续物种注释的一个假阴性问题。因此,在本公开中,通过引入步骤208,可实现对病原体基因组的回捞,从而可以使得所构建的微生物基因组数据库的覆盖范围包括具有临床意义的病原体微生物,进而有助于保证所构建的数据库的整体质量,减少假阴性的检索结果,进而提升该数据库在病原体检测方面的应用性。
48.应了解,在本公开中,基于所标识出的基因组数据以及该第一基因组数据分组确定经最终筛选的基因组数据指的是经最终筛选的基因组数据由所标识出的基因组数据以及该第一基因组数据分组组成。
49.在步骤210,对经最终筛选的基因组数据进行去冗余处理,以便得到用于构建微生物基因组数据库的基因组数据。
50.在本公开中,通过对经最终筛选的基因组数据进行去冗余处理可有效地降低所构建的数据库的数据量,同时还可保持其所包括的基因组的多样性,进而有助于提高基于该数据库进行微生物物种鉴定的效率,而不会降低其准确性。
51.下面将结合图4对步骤210作进一步更详细的描述。
52.在一些实施例中,在对经筛选的基因组数据进行去冗余处理之前,还包括从所述经筛选的基因组数据中去除包括与质粒序列相关联的基因组数据。
53.图3示出了根据本公开的实施例的用于对所获取的基因组数据进行初步筛选的方法300的流程图。方法300可由如图1所示的计算设备110执行,也可以在图6所示的电子设备600处执行。应当理解的是,方法300还可以包括未示出的附加框和/或可以省略所示出的框,本公开的范围在此方面不受限制。
54.在步骤302,从所获取的基因组数据所涉及的多个微生物类别中标识出在物种分类系统中分类有误(包括分类不明确)的一个或多个微生物类别。
55.此处,所获取的基因组数据为在步骤202中获取的基因组数据。
56.在物种分类系统中,以结构树的形式给出了当前已知的每一生物物种(包括微生物物种)的类别(例如属于哪个科、哪个目和哪个属等),但是新出现的物种在物种分类系统中可能还没有被明确分类(即是未经分类的),由此可认为这些物种的分类是待定的(即不明确的),也可认为其分类是有误的,如果将这样的微生物物种也归入了待构建的数据库中,将影响该数据库的精准性。
57.在步骤304,从所获取的基因组数据中移除与所标识出的一个或多个微生物类别相关联的基因组数据,以便得到经初步筛选的基因组数据。
58.在本公开中,通过将与被错误分类的一个或多个微生物类别的基因组数据移除,可确保所构建的数据库中所包括的数据都是被正确分类的基因组数据,从而有助于提高待构建的微生物基因组数据库的准确性。
59.图4示出了示出了根据本公开的实施例的用于对经筛选的基因组数据进行去冗余处理的方法400的流程图。方法400可由如图1所示的计算设备110执行,也可以在图6所示的电子设备600处执行。应当理解的是,方法400还可以包括未示出的附加框和/或可以省略所示出的框,本公开的范围在此方面不受限制。
60.在步骤402,对于任一微生物物种,如果在经最终筛选的基因组数据中包括与该微生物物种的两个或更多个基因组相关联的基因组数据,则从该两个或更多个基因组中确定该微生物物种的基础基因组。
61.通常情况下,基于公共数据库获得的基因组数据中,一个微生物物种都包括两个或更多基因组,例如以大肠杆菌为例,其可包括例如2886个质控后的菌株基因组。
62.在本公开中,基础基因组应该是该两个或更多个基因组中物种组装水平相对较好并且有文献支持的基因组。下面将基于图5对步骤402作进一步更详细的描述。
63.在步骤404,基于该两个或更多个基因组中除该基础基因组以外的每一基因组的片段序列,根据预定长度和预定步长将相应基因组打断成多个片段序列。在本公开中,通过打断得到的每一片段序列的长度等于该预定长度。
64.在一些实施例中,该预定长度大于等于10bp(即10碱基对)并且小于等于300bp,并且该预定步长大于等于1bp。
65.步长指的是每次打断时,需要移动的距离的长度。例如,在预定长度为20bp,预定步长待1bp的实施例中,打断得到的第一片段序列是由从第0bp到第20bp的数据组成的序列,打断得到的第二片段序列是从第1bp到第21bp的数据组成的序列,打断得到的第三片段序列是从第2bp到第22bp的数据组成的序列,并以此类推。
66.在步骤406,将分割得到的每一片段序列与基础基因组进行比对,以便确定该片段序列是否为与基础基因组的相似度小于预定阈值的特异性序列。
67.在本公开中,步骤406中提到的预定阈值大于0并且小于1,优选地大于0.5并且小于1,例如为0.7。
68.例如仅作为示例,假设该预定阈值为0.7,如果在基础基因组中找到了与相应的片段序列完全相同的部分,则确定该片段序列与该基础基因组的相似度为100%(即为1),因此该片段序列不属于特异性序列。
69.又如,同样假设该预定阈值为0.7,如果在基础基因组中找到了与相应的片段序列的相似度为40%的部分,则确定该片段序列与该基础基因组的相似度为40%(即为0.4),因此该片段序列属于特异性序列。
70.在步骤408,基于为该微生物物种确定的特异性序列以及基础基因组,确定该微生物物种的完整基因组。
71.在本公开中,对于任何微生物物种,可通过将为该微生物物种确定的各个特异性序列和集成基因组组合在一起,就可确定该微生物物种的完整基因组。
72.在本公开中,微生物物种的完整基因组指的是该微生物物种的基本上完整的基因组,其基本上可以反映该微生物物种的一套染色体的完整的dna序列。
73.具体地,可通过以下方式来确定该微生物物种的完整基因组。
74.首先,将为该微生物物种确定的所有特异性序列中具有连续性特征的多个特异性序列进行组装,以便得到多个经组装的特异性序列。
75.然后,将多个经组装的特异性序列(即在前面一个步骤得到的各个经组装的特异性序列)根据长度进行排序,以便将这些经组装的特异性序列中排名靠后的多个经组装的特异性序列丢弃,其中被丢弃的多个经组装的特异性序列的长度之和小于或等于所有特异性序列的总长度的预定比例(例如,10%)。
76.然后,以任意顺序将剩下的经组装的特异性序列和基础基因组整合成所述微生物物种的完整基因组。
77.在一些实施例中,可通过将剩余的经组装的特异性序列全部置于基础基因组的最前面或者全部至于基础基因组的最后面来得到完整基因组。
78.在步骤410,将与完整基因组相关联的基因组数据保存到微生物基因组数据库中,作为微生物物种的基因组数据。
79.在本公开中,通过以上方式,可以很好的解决去冗余问题,大大地减少所创建的微生物基因组数据库中与各个微生物物种相关联的基因组数据的数据量(例如,以大肠杆菌为例,可包括2886个质控后的菌株基因组,这些基因组的大小一共为14g,但使用本公开的以上方法400进行冗余处理后所得到的基因组大小仅为60m),而仍可保留各个微生物物种的基因组的完整性和多样性,进而有助于提高微生物物种鉴定时的比对效率。
80.图5示出了示出了根据本公开的实施例的用于从两个或更多个基因组中确定微生物物种的基础基因组的方法500的流程图。方法500是对步骤402的实现实施例,其可由如图1所示的计算设备110执行,也可以在图6所示的电子设备600处执行。应当理解的是,方法500还可以包括未示出的附加框和/或可以省略所示出的框,本公开的范围在此方面不受限制。
81.在步骤502,确定两个或更多个基因组(即,当前进行判断的微生物物种的两个或更多个基因组)中的每一基因组的组装水平。
82.在步骤504,从该两个或更多个基因组中标识出组装水平的级别最高的一个或多个基因组。
83.在本公开中,级别最高的组装水平为例如前面提到的完整基因组(complete genome)。
84.在步骤506,确定所标识的一个或多个基因组的文献支持数量。
85.在本公开中,步骤506中提到的基因组的文献支持数量指的是利用爬虫技术在生物医学文献数据库中对该基因组进行批量检索所检索到的文献的数量。
86.在步骤508,将所标识的一个或多个基因组中具有最大文献支持数量的基因组确定为该微生物物种的基础基因组。
87.应了解,在本公开中,如果所标识的一个或多个基因组中具有最大文献支持数量的基因组的数量为两个或更多个,则可在其中任取一个基因组作为相应微生物物种的基础基因组。
88.下面以具有2886个基因组的大肠杆菌为例对本公开所能实现的效果进行说明。
89.通过实验发现,如果将大肠杆菌的该2886个基因组全部模拟成下机数据,并随机抽取10万条读序(reads),基于本公开构建的微生物基因组数据库的比对率可达到96.37%,所需的比对时间为3.265秒,因此其比对效率(即分析效率)较高,能够快速完成一次数据分析。应了解,比对率越高说明数据库中涵盖该物种菌株的多样性越大(即,可以检出该物种更多的菌株)。对于现有技术中的单一基因组数据库(即,以基础基因组为物种代表基因组构建数据库),由于其没有纳入其他菌株基因组信息,但在测序时如果样本中为非基础基因组菌株则很可能造成假阴性问题,因此基于该数据库的比对率可达83.807%,所需的比对时间为2.463秒。对于现有技术中的所有基因组融合数据库(即所有基因组经融合
处理的数据库),由于其组装过程中不可避免的会丢掉其他菌株的特异性序列,而且较为依赖组装软件的性能,因此基于该数据库的比对率仅为39.232%,所需的比对时间为3.119秒。对于现有技术中的所有基因组未经任何处理的数据库,由于没有对基因组进行任何影响特异性序列丢失的操作,因此基于该数据库的比对率可达100%,但是由于数据量大,其所需的比对时间却长达50.569秒,因此其比对效率(即分析效率)明显低于其他几个数据库。
90.由此可见,本公开构建的微生物基因组数据库的比对率接近现有技术中的所有基因组未经任何处理的数据库,而且所用的时间接近单一基因组数据库以及全部基因组融合数据库,因此本公开的微生物基因组数据库同时具有较高的比对率和比对效率,其效果明显好于现有的数据库。
91.图6示出了可以用来实施本公开内容的实施例的示例电子设备600的示意性框图。例如,如图1所示的计算设备110可以由电子设备600来实施。如图所示,电子设备600包括中央处理单元(cpu)601,其可以根据存储在只读存储器(rom)602中的计算机程序指令或者从存储单元608加载到随机存取存储器(ram)603中的计算机程序指令,来执行各种适当的动作和处理。在随机存取存储器603中,还可存储电子设备600操作所需的各种程序和数据。中央处理单元601、只读存储器602以及随机存取存储器603通过总线604彼此相连。输入/输出(i/o)接口605也连接至总线604。
92.电子设备600中的多个部件连接至输入/输出接口605,包括:输入单元606,例如键盘、鼠标、麦克风等;输出单元607,例如各种类型的显示器、扬声器等;存储单元608,例如磁盘、光盘等;以及通信单元609,例如网卡、调制解调器、无线通信收发机等。通信单元609允许设备600通过诸如因特网的计算机网络和/或各种电信网络与其他设备交换信息/数据。
93.上文所描述的各个过程和处理,例如方法200-500,可由中央处理单元601执行。例如,在一些实施例中,方法200-500可被实现为计算机软件程序,其被有形地包含于机器可读介质,例如存储单元608。在一些实施例中,计算机程序的部分或者全部可以经由只读存储器602和/或通信单元609而被载入和/或安装到设备600上。当计算机程序被加载到随机存取存储器603并由中央处理单元601执行时,可以执行上文描述的方法200-500的一个或多个动作。
94.本公开涉及方法、装置、系统、电子设备、计算机可读存储介质和/或计算机程序产品。计算机程序产品可以包括用于执行本公开的各个方面的计算机可读程序指令。
95.计算机可读存储介质可以是可以保持和存储由指令执行设备使用的指令的有形设备。计算机可读存储介质例如可以是――但不限于――电存储设备、磁存储设备、光存储设备、电磁存储设备、半导体存储设备或者上述的任意合适的组合。计算机可读存储介质的更具体的例子(非穷举的列表)包括:便携式计算机盘、硬盘、随机存取存储器(ram)、只读存储器(rom)、可擦式可编程只读存储器(eprom或闪存)、静态随机存取存储器(sram)、便携式压缩盘只读存储器(cd-rom)、数字多功能盘(dvd)、记忆棒、软盘、机械编码设备、例如其上存储有指令的打孔卡或凹槽内凸起结构、以及上述的任意合适的组合。这里所使用的计算机可读存储介质不被解释为瞬时信号本身,诸如无线电波或者其他自由传播的电磁波、通过波导或其他传输媒介传播的电磁波(例如,通过光纤电缆的光脉冲)、或者通过电线传输的电信号。
96.这里所描述的计算机可读程序指令可以从计算机可读存储介质下载到各个计算/处理设备,或者通过网络、例如因特网、局域网、广域网和/或无线网下载到外部计算机或外部存储设备。网络可以包括铜传输电缆、光纤传输、无线传输、路由器、防火墙、交换机、网关计算机和/或边缘计算设备。每个计算/处理设备中的网络适配卡或者网络接口从网络接收计算机可读程序指令,并转发该计算机可读程序指令,以供存储在各个计算/处理设备中的计算机可读存储介质中。
97.用于执行本公开操作的计算机程序指令可以是汇编指令、指令集架构(isa)指令、机器指令、机器相关指令、微代码、固件指令、状态设置数据、或者以一种或多种编程语言的任意组合编写的源代码或目标代码,所述编程语言包括面向对象的编程语言—诸如smalltalk、c 等,以及常规的过程式编程语言—诸如“c”语言或类似的编程语言。计算机可读程序指令可以完全地在用户计算机上执行、部分地在用户计算机上执行、作为一个独立的软件包执行、部分在用户计算机上部分在远程计算机上执行、或者完全在远程计算机或服务器上执行。在涉及远程计算机的情形中,远程计算机可以通过任意种类的网络—包括局域网(lan)或广域网(wan)—连接到用户计算机,或者,可以连接到外部计算机(例如利用因特网服务提供商来通过因特网连接)。在一些实施例中,通过利用计算机可读程序指令的状态信息来个性化定制电子电路,例如可编程逻辑电路、现场可编程门阵列(fpga)或可编程逻辑阵列(pla),该电子电路可以执行计算机可读程序指令,从而实现本公开的各个方面。
98.这里参照根据本公开实施例的方法、装置(系统)和计算机程序产品的流程图和/或框图描述了本公开的各个方面。应当理解,流程图和/或框图的每个方框以及流程图和/或框图中各方框的组合,都可以由计算机可读程序指令实现。
99.这些计算机可读程序指令可以提供给通用计算机、专用计算机或其它可编程数据处理装置的处理单元,从而生产出一种机器,使得这些指令在通过计算机或其它可编程数据处理装置的处理单元执行时,产生了实现流程图和/或框图中的一个或多个方框中规定的功能/动作的装置。也可以把这些计算机可读程序指令存储在计算机可读存储介质中,这些指令使得计算机、可编程数据处理装置和/或其他设备以特定方式工作,从而,存储有指令的计算机可读介质则包括一个制造品,其包括实现流程图和/或框图中的一个或多个方框中规定的功能/动作的各个方面的指令。
100.也可以把计算机可读程序指令加载到计算机、其它可编程数据处理装置、或其它设备上,使得在计算机、其它可编程数据处理装置或其它设备上执行一系列操作步骤,以产生计算机实现的过程,从而使得在计算机、其它可编程数据处理装置、或其它设备上执行的指令实现流程图和/或框图中的一个或多个方框中规定的功能/动作。
101.附图中的流程图和框图显示了根据本公开的多个实施例的系统、方法和计算机程序产品的可能实现的体系架构、功能和操作。在这点上,流程图或框图中的每个方框可以代表一个模块、程序段或指令的一部分,所述模块、程序段或指令的一部分包含一个或多个用于实现规定的逻辑功能的可执行指令。在有些作为替换的实现中,方框中所标注的功能也可以以不同于附图中所标注的顺序发生。例如,两个连续的方框实际上可以基本并行地执行,它们有时也可以按相反的顺序执行,这依所涉及的功能而定。也要注意的是,框图和/或流程图中的每个方框、以及框图和/或流程图中的方框的组合,可以用执行规定的功能或动
作的专用的基于硬件的系统来实现,或者可以用专用硬件与计算机指令的组合来实现。
102.以上已经描述了本公开的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。本文中所用术语的选择,旨在最好地解释各实施例的原理、实际应用或对市场中的技术改进,或者使本技术领域的其它普通技术人员能理解本文披露的各实施例。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。