1.本发明属于化工技术领域,特别是涉及一种4位氨基取代咔唑、二苯并[b,d]呋喃和芴衍生物的合成方法。
背景技术:
[0002]
咔唑、二苯并[b,d]呋喃和芴衍生物作为最为重要的杂环骨架,在传统工业中常用于生产燃料、光电导体、感光材料、特种油墨等,具有耐紫外光、热稳定性好的特点,特别是咔唑在其中作用巨大,近年来,越来越多的工作表明4-氨基咔唑、二苯并[b,d]呋喃和芴衍生物具备独特的生物活性和更为优秀的光电物理性质。
[0003]
一直以来咔唑及其衍生物的c-h官能化局限在1位和3位,这是由于咔唑骨架的氨基是给电子基团,其亲电取代反应具有邻对位位置选择性,然而,4-氨基咔唑、二苯并[b,d]呋喃和芴衍生物的合成方法发展缓慢,目前的主流合成方法需要四步合成,其中两步需要用到钯催化偶联反应,成本高昂且底物局限性大:a)以邻溴苯硼酸为原料在钯催化下与2-硝基溴苯发生suzuki-miyaura偶联生成2-溴-2'-硝基-1,1'-联苯;b)2-溴-2'-硝基-1,1'-联苯在三苯基膦的二氯苯溶液中回流得到4-溴咔唑;c)在咔唑的氮上上保护剂团;d)在钯催化下三叔丁基膦作为配体通过buchwald
–
hartwig偶联得到4-氨基咔唑,整个过程的反应条件苛刻,并且难以合成高官能团化的4-氨基咔唑,也无法得到其他相似骨架,因此,一步合成4-氨基咔唑、二苯并[b,d]呋喃和芴衍生物一直是一个尚未解决的问题,并引起了人们的广泛关注。
技术实现要素:
[0004]
本发明的目的在于提供一种4位氨基取代咔唑、二苯并[b,d]呋喃和芴衍生物的合成方法,解决了现有的4-氨基咔唑、二苯并[b,d]呋喃和芴衍生物合成步骤繁琐、成本高、收率低的技术问题。
[0005]
为达上述目的,本发明是通过以下技术方案实现的:
[0006]
一种4位氨基取代咔唑、二苯并[b,d]呋喃和芴衍生物的合成方法,包括如下步骤:
[0007]
将邻n-苯基卤代苯胺或邻苯氧基卤代苯或邻苄基卤代苯为起始原料,将胺类衍生物和降冰片烯衍生物为原料,加入到反应容器中;
[0008]
使用钯及其含钯的金属盐作为催化剂,并加入添加剂、配体和碱,升温至60-150摄氏度;
[0009]
检测原料反应完,蒸馏除去溶剂、柱层析或其他分离手段得到4位氨基取代咔唑或二苯并[b,d]呋喃或芴衍生物。
[0010]
可选的,钯催化剂为醋酸钯、氯化钯、新戊酸钯、钯碳、金属钯、二三苯基膦二氯化钯、四三苯基膦钯等常用钯催化剂中的一种,添加剂为羧酸、金属盐或相转移催化剂。
[0011]
可选的,碱为碳酸铯、碳酸钾、碳酸钠、磷酸钾、磷酸钠、三乙胺等常用有机无机碱
中的一种,溶剂为乙腈、二氯甲烷、四氢呋喃、二氧六环、二甲基甲酰胺、硝基甲烷、甲苯或二氯乙烷等常用溶剂中的一种。
[0012]
可选的,配体为三苯基膦类衍生物、双齿膦配体、buchwald配体类衍生物、卡宾类配体等常用配体中的一种。
[0013]
本发明的实施例具有以下有益效果:
[0014]
本发明的一个实施例通过此方法一步构建4位氨基取代咔唑或二苯并[b,d]呋喃或芴衍生物,产物可以用于药物的合成,且具有产率高、成本低、操作步骤简便的特点,减少溶剂需要进一步处理的情况。
[0015]
当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有优点。
附图说明
[0016]
构成本技术的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0017]
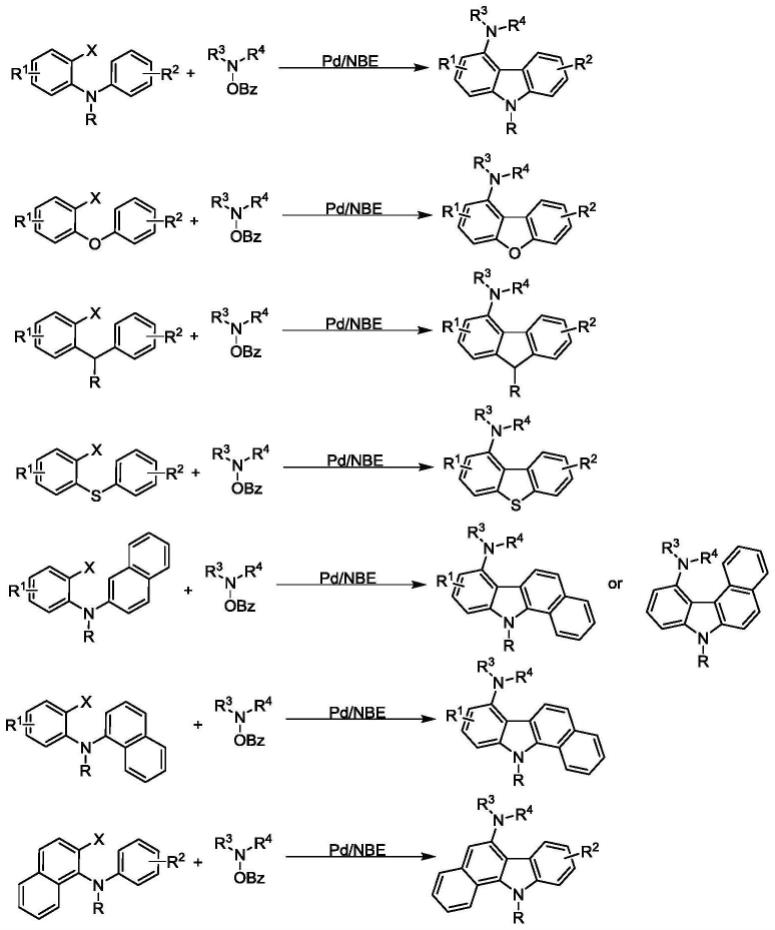
图1为本发明一实施例的反应方程示意图;
[0018]
图2为本发明一实施例的n-甲酸甲酯-n-苯基邻碘苯胺反应示意图;
[0019]
图3为本发明一实施例的2-碘-n-甲基-n-苯基苯胺反应示意图;
[0020]
图4为本发明一实施例的1-碘-2-苯氧基苯反应示意图。
具体实施方式
[0021]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。以下对至少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使用的任何限制。
[0022]
为了保持本发明实施例的以下说明清楚且简明,本发明省略了已知功能和已知部件的详细说明。
[0023]
请参阅图1-4所示,在本实施例中提供了一种4位氨基取代咔唑、二苯并[b,d]呋喃和芴衍生物的合成方法,包括如下步骤:
[0024]
将邻n-苯基卤代苯胺或邻苯氧基卤代苯或邻苄基卤代苯为起始原料(基团x为碘i、溴br、氯cl、三氟甲磺酸基otf等),将胺类衍生物(基团x’为obz、cl、ots等类似离去基团)和降冰片烯衍生物为原料,加入到反应容器中;
[0025]
使用钯及其含钯的金属盐作为催化剂,并加入添加剂、配体和碱,升温至60-150摄氏度;
[0026]
检测原料反应完,蒸馏除去溶剂、柱层析或其他分离手段得到4位氨基取代咔唑或二苯并[b,d]呋喃或芴衍生物,柱层析时使用的流动相的组成和体积比为石油醚:乙酸乙酯=10:1-50:1;基团r为烷基、烯基、硝基、酰基、卤素(br,cl)、烷氧基、氧酯基、炔烃、芳基、氢等分子基团;基团r1和r2为烷基、烯基、硝基、酰基、卤素(br,cl)、烷氧基、氧酯基、炔烃、芳基等分子基团;芳烃上可有单个或多个不同r1和r2基团,也可为苯并环,例如稠环或烷基化等;r3、r4可为烷基(伯碳、仲碳、叔碳)等基团。
[0027]
本实施例一个方面的应用为:将n-苯基卤代苯胺(也可为邻苯氧基卤代苯或邻苄
基卤代苯)、胺类衍生物和降冰片烯类衍生物加到反应容器中,加入钯催化剂、添加剂、配体、碱,排去反应容器中空气,并替换为惰性气体(如氩气,氮气等)后密封反应容器,升温至60-150摄氏度,使用薄层色谱、气相色谱等检测原料反应进度,待反应原料消失后过滤,用乙酸乙酯洗涤并收集滤液,减压浓缩后,再进行蒸馏,并柱层析分离,得到4位氨基取代咔唑(根据底物不同可得到二苯并[b,d]呋喃或芴衍生物)。
[0028]
通过此方法一步构建4位氨基取代咔唑或二苯并[b,d]呋喃或芴衍生物,产物可以用于药物的合成,且具有产率高、成本低、操作步骤简便的特点,减少溶剂需要进一步处理的情况。
[0029]
如图1所示,本实施例的溶剂为乙腈、二氯甲烷、四氢呋喃、二氧六环、二甲基甲酰胺、硝基甲烷、甲苯或二氯乙烷等常用溶剂中的一种。钯催化剂为醋酸钯、氯化钯、新戊酸钯、钯碳、金属钯、二三苯基膦二氯化钯、四三苯基膦钯等常用钯催化剂中的一种,钯催化剂与n-苯基卤代苯胺物质的量比为1:10到1:20。配体为三苯基膦类衍生物、双齿膦配体、buchwald配体类衍生物、卡宾类配体等常用配体中的一种,钯催化剂与配体物质的量的比为1:2。
[0030]
如图1所示,本实施例的碱为碳酸铯、碳酸钾、碳酸钠、磷酸钾、磷酸钠、三乙胺等常用有机无机碱中的一种,碱与胺衍生物物质的量比为4:1。添加剂为羧酸、金属盐或相转移催化剂,例如特戊酸、苯甲酸、乙酸、碘化钠、冠醚等,添加剂与n-苯基卤代苯胺物质的量比为2:5。
[0031]
实施例1:在25ml反应管中,添加n-甲酸甲酯-n-苯基邻碘苯胺(0.2mmol,70.6mg)、苯甲氧基吗啉(0.4mmol,82.8mg)、醋酸钯(0.02mmol,4.5mg)、三苯基膦(0.04mmol,10.5mg)、碳酸铯(0.8mmol,260mg)、特戊酸(0.08mmol,8.2mg),然后在氩气氛围下加入甲苯(3ml)和降冰片烯(0.8mmol,75mg),用盖子密封,将所得的淡黄色悬浮液在室温下搅拌15分钟,然后置于120℃预加热的油浴中,在900-1200转/分钟搅拌12小时,反应结束后,用硅胶色谱柱(石油醚:乙酸乙酯=10:1)纯化残留物,最终得到白色固体(41.5mg,产率67.0%);
[0032]
产物检测数据如下:
[0033]1h nmr(400mhz,chloroform-d)δ8.35
–
8.26(m,2h),8.08(d,j=8.3hz,1h),7.49
–
7.36(m,3h),7.05(d,j=7.9hz,1h),4.12(s,3h),4.02(s,4h),3.28
–
3.17(m,2h),3.02(s,2h);
[0034]
13
c nmr(151mhz,chloroform-d)δ152.82,148.54,139.64,137.85,127.71,126.42,125.04,123.29,122.51,119.11,115.75,112.93,111.59,67.26,53.51,52.23;
[0035]
hrms(esi)m/z:[m h]
calcd for c
18h19
n2o
3 311.1390;found 311.1389。
[0036]
实施例2:在25ml烧瓶中,添加2-碘-n-甲基-n-苯基苯胺(0.2mmol,61.8mg)、苯甲氧基哌嗪(0.4mmol,122.8mg)、醋酸钯(0.02mmol,4.5mg)、三苯基膦(0.04mmol,10.5mg)、碳酸铯(0.8mmol,260mg)、特戊酸(0.08mmol,8.2mg),然后在氩气氛围下加入甲苯(3ml)和降冰片烯(0.8mmol,75mg),用盖子密封,将所得的淡黄色悬浮液在室温下搅拌15分钟,然后置于120℃预加热的油浴中,在900-1200转/分钟搅12小时,反应结束后,用硅胶色谱柱(石油醚:乙酸乙酯=10:1)纯化残留物,最终得到白色固体(63.0mg,产率86%);
[0037]
产物检测数据如下:
[0038]1h nmr(600mhz,chloroform-d)δ8.22(d,j=7.8hz,1h),7.49
–
7.44(m,1h),7.43
–
7.37(m,2h),7.25(t,j=7.5hz,1h),7.14(d,j=8.1hz,1h),6.85(d,j=7.7hz,1h),4.16(s,2h),3.82(s,3h),3.41(s,4h),2.88(s,2h),1.51(s,9h);
[0039]
13
c nmr(151mhz,chloroform-d)δ154.97,148.91,142.40,140.55,126.30,124.92,122.60,121.75,118.96,115.72,108.14,108.06,104.03,79.77,51.63,43.87,29.16,28.45;
[0040]
hrms(esi)m/z:[m h]
calcd for c
22h28
n3o
2 366.2176;found 366.2168。
[0041]
实施例3:在25ml烧瓶中,添加1-碘-2-苯氧基苯(0.2mmol,59.2mg)、苯甲氧基吗啉(0.4mmol,82.8mg)、醋酸钯(0.02mmol,4.5mg)、三苯基膦(0.04mmol,10.5mg)、碳酸铯(0.8mmol,260mg)、特戊酸(0.08mmol,8.2mg),然后在氩气氛围下加入甲苯(3ml)和降冰片烯(0.8mmol,75mg),用盖子密封,将所得的淡黄色悬浮液在室温下搅拌15分钟,然后置于120℃预加热的油浴中,在900~1200转/分钟搅拌12小时,反应结束后,用硅胶色谱柱(石油醚/乙酸乙酯=10:1)纯化残留物,最终得到无色油状液体(21.3mg,产率42.0%);
[0042]
产物检测数据如下:
[0043]1h nmr(400mhz,chloroform-d)δ7.95(d,j=7.7hz,1h),7.62
–
7.55(m,1h),7.48
–
7.34(m,3h),7.31
–
7.24(m,1h),6.94(d,j=7.9hz,1h),4.05
–
4.02(m,4h),3.22
–
3.18(m,4h);
[0044]
13
c nmr(151mhz,chloroform-d)δ157.36,155.74,148.78,127.83,126.34,123.32,122.73,122.54,117.22,111.30,111.20,106.65,67.26,51.81;
[0045]
hrms(esi)m/z:[m h]
calcd for c
16h16
no
2 254.1176;found 254.1168。
[0046]
上述实施例可以相互结合。
[0047]
需要说明的是,本技术的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本技术的实施方式能够以除了在这里图示或描述的那些以外的顺序实施。
[0048]
在本发明的描述中,需要理解的是,方位词如“前、后、上、下、左、右”、“横向、竖向、垂直、水平”和“顶、底”等所指示的方位或位置关系通常是基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,在未作相反说明的情况下,这些方位词并不指示和暗示所指的装置或元件必须具有特定的方位或者以特定的方位构造和操作,因此不能理解为对本发明保护范围的限制;方位词“内、外”是指相对于各部件本身的轮廓的内外。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。