1.本发明属于胶黏剂领域,具体涉及一种双组分环氧胶黏剂及其制备方法和应用。
背景技术:
2.在太阳能光伏制造过程中,主要工序是将体积比较大的硅块利用线切割工艺切割成非常薄(200μm以下)的硅片。在硅块的切割工艺中,先用胶黏剂将可以重复使用的钢板与一次性使用的10mm以上的厚基板(玻璃板、石墨板或树脂板,下同)粘接,然后再用胶黏剂将硅块和一次性使用的厚基板粘接,之后便可用线切割机进行后续的硅块切割。切割完成后,硅块变成了硅片,10mm以上厚的基板,因为切割钢线有着一定的切割幅度,所以也会被切割数毫米的深度,故基板为一次性使用材料。钢板未被切割,可以重复使用。在每次使用钢板之前,均需要将粘接在钢板上的基板从表面完全清除。
3.特殊用途光伏切割胶黏剂具有巨大的市场跟生存空间,产品不同于其他胶黏剂,不仅要求具有快干、高强度且在切割过程中耐溶剂及不同化学药品侵蚀,而且又要求在特定条件下具有清理性。目前常用的方法是通过加热的方式清胶,具体地,采用150℃以上的烘箱或者热台,通过长时间热老化将胶黏剂及厚基板脱落清除。然而,该方法浪费能源,需借助人力,清理效率低下,并且在清胶过程中还会有挥发性气体产生。此外,也可以通过水煮(或者酸碱煮)的方式清胶,胶水煮温度一般为90-100℃,传统的双组分胶黏剂所需水煮脱胶时间较长,因此会影响生产效率并浪费能源。
技术实现要素:
4.本发明的目的是为了克服现有的光伏切割胶黏剂使用后清胶效率较低的缺陷,而提供一种新的双组分环氧胶黏剂及其制备方法和应用,该双组分环氧胶黏剂经水煮后能够快速脱胶。
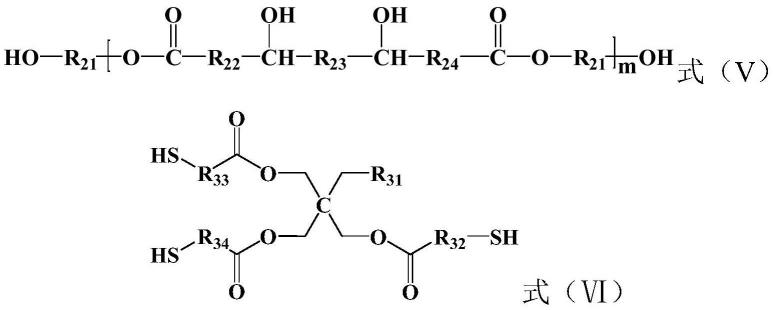
5.本发明的第一方面提供了一种双组分环氧胶黏剂,所述双组分环氧胶黏剂含有组分a和组分b,所述组分a中含有环氧树脂30-70重量份、第一填料20-60重量份、第一发泡剂5-30重量份和防沉降剂0.5-2重量份,所述组分b中含有聚氨酯型固化剂30-60重量份、含叔胺催化剂20-50重量份、第二填料10-40重量份、第二发泡剂1-10重量份和膨胀剂1-15重量份;所述聚氨酯型固化剂具有式(i)所示的结构:
[0006][0007]
式(i)中,r1为c
1-c
10
的亚烷基、c
6-c
20
的亚环烷基或c
6-c
20
的亚芳基,r2为衍生自亲水性低聚物多元醇的基团,r3为带有至少两个巯基的c
1-c
30
的巯基烷基或c
1-c
30
的巯基酯基,n为大于等于1的整数。
[0008]
在一种优选实施方式中,r2具有式(ii)所示的结构:
[0009][0010]
式(ii)中,r
21
为c
1-c
10
的亚烷基,r
22
、r
23
和r
24
各自独立地为c
1-c5的亚烷基或不存在,m为大于等于1的整数。
[0011]
在一种优选实施方式中,r3具有式(iii)所示的结构:
[0012][0013]
式(iii)中,r
31
为c
1-c5的烷基或-o-co-r
31-sh,r
31
为c
1-c5的亚烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。
[0014]
在一种优选实施方式中,所述聚氨酯型固化剂在25℃下的粘度为3000-30000cps。
[0015]
在一种优选实施方式中,所述聚氨酯型固化剂按照以下方法制备得到:
[0016]
s1、在惰性气氛下,将式(iv)所示的多异氰酸酯与式(v)所示的亲水性低聚物多元醇进行聚加成反应,得到仅裸露异氰酸酯基团在分子链末端的α,ω-双异氰酸酯聚氨酯前体;
[0017]
s2、在惰性气氛下,将步骤s1中所得α,ω-双异氰酸酯聚氨酯前体与式(ⅵ)所示的含巯基化合物在催化剂的作用下进行聚加成反应,得到聚氨酯型固化剂;
[0018]
ocn-r
1-nco
ꢀꢀ
(式iv)
[0019][0020]
式(iv)中,r1为c
1-c
10
的亚烷基、c
6-c
20
的亚环烷基或c
6-c
20
的亚芳基;
[0021]
式(v)中,r
21
为c
1-c
10
的亚烷基,r
22
、r
23
和r
24
各自独立地为c
1-c5的亚烷基或不存在,m为大于等于1的整数;
[0022]
式(ⅵ)中,r
31
为c
1-c5的烷基或-o-co-r
31-sh,r
31
为c
1-c5的亚烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。
[0023]
在一种优选实施方式中,步骤s1中,所述聚加成反应的条件包括温度为90-120℃,时间为2-5h。
[0024]
在一种优选实施方式中,步骤s2中,所述聚加成反应的条件包括温度为20-50℃,时间为2-10h。
[0025]
在一种优选实施方式中,步骤s1中,所述多异氰酸酯与亲水性低聚物多元醇的摩尔比为1:(0.6-0.8)。
[0026]
在一种优选实施方式中,步骤s2中,所述多异氰酸酯与含巯基化合物的摩尔比为1:(0.2-1)。
[0027]
在一种优选实施方式中,步骤s2中,所述催化剂的用量为单体总质量的0.1wt%-0.3wt%。
[0028]
在一种优选实施方式中,步骤s1中,所述亲水性低聚物多元醇通过将式(
ⅷ
)所示的二元醇与式(
ⅸ
)所示的二元羧酸进行缩合反应得到;
[0029]
ho-r
21-oh
ꢀꢀ
式(viii)
[0030][0031]
式(
ⅷ
)中,r
21
为c
1-c
10
的亚烷基;
[0032]
式(
ⅸ
)中,r
22
、r
23
和r
24
各自独立地为c
1-c5的亚烷基或不存在。
[0033]
在一种优选实施方式中,所述组分a和组分b的质量混合比为1:(1-1.3)。
[0034]
在一种优选实施方式中,所述环氧树脂为双酚a环氧树脂和/或双酚f环氧树脂。
[0035]
在一种优选实施方式中,所述第一填料和第二填料各自独立地选自滑石粉、钛白粉、高岭土、碳酸钙、硫酸钡以及氧化锌中的至少一种。
[0036]
在一种优选实施方式中,所述第一发泡剂和第二发泡剂各自独立地选自碳酸氢钙、碳酸铵、偶氮二异丁腈、偶氮二甲酸二乙酯、对苯磺酰肼以及4,4
’‑
二磺酰肼二苯醚中的至少一种。
[0037]
在一种优选实施方式中,所述防沉降剂为有机膨润土。
[0038]
在一种优选实施方式中,所述含叔胺催化剂为改性脂肪叔胺。
[0039]
在一种优选实施方式中,所述膨胀剂为钙矾石。
[0040]
本发明的第二方面提供了上述双组分环氧胶黏剂的制备方法,该方法包括以下步骤:
[0041]
(1)将环氧树脂、第一填料、第一发泡剂和防沉降剂按比例混合均匀,抽真空除泡后,得到组分a;
[0042]
(2)将聚氨酯型固化剂、含叔胺催化剂、第二填料、第二发泡剂和膨胀剂按比例混合均匀,抽真空除泡后,得到组分b;
[0043]
(3)将组分a和组分b混合均匀,得到双组分环氧胶黏剂。
[0044]
本发明的第三方面提供了所述双组分环氧胶黏剂作为光伏切割胶黏剂的应用。
[0045]
本发明的关键在于以具有式(i)所示结构的聚氨酯型化合物作为固化剂,同时辅以发泡剂和膨胀剂的使用,由此所得双组分环氧胶黏剂在水煮环境下可以快速脱胶,提高生产效率,节约能源,将其作为光伏切割用胶黏剂时能够显著提高清胶效率。推测其原因,可能是由于:一方面,具有式(i)所示结构的聚氨酯型固化剂以聚氨酯作为主链且侧链上含有大量羟基,这种特定的结构能够提高胶膜的亲水性,有利于胶层在水煮环境中快速脱落;另一方面,膨胀剂的使用能够在水煮过程中帮助固化后的胶体膨胀,增加与水的接触面积,同时在水煮环境下,这种特定的聚氨酯型固化剂能够在膨胀剂的辅助作用下作为输水介质
将水分传递至胶层内的发泡剂,使得水煮后的胶层均匀发泡,如此有利于胶层快速完整脱落,钢板无需清理残胶即可回收,有利于提高清胶效率。
具体实施方式
[0046]
本发明提供的双组分环氧胶黏剂含有组分a和组分b。其中,所述组分a和组分b的质量混合比优选为1:(1-1.3),如1:1.0、1:1.1、1:1.2、1:1.3等。
[0047]
所述组分a中含有环氧树脂、第一填料、第一发泡剂和防沉降剂。其中,所述环氧树脂的含量为30-70重量份,如30、35、40、45、50、55、60、65、70重量份等;所述第一填料的含量为20-60重量份,如20、25、30、35、40、45、50、55、60重量份等;所述第一发泡剂的含量为5-30重量份,如5、10、15、20、25、30重量份等;所述防沉降剂的含量为0.5-2重量份,如0.5、0.8、1、1.2、1.5、1.8、2重量份等。
[0048]
所述组分b中含有聚氨酯型固化剂、含叔胺催化剂、第二填料、第二发泡剂和膨胀剂。其中,所述聚氨酯型固化剂的含量为30-60重量份,如30、35、40、45、50、55、60重量份等;所述含叔胺催化剂的含量为20-50重量份,如20、25、30、35、40、45、50重量份等;所述第二填料的含量为10-40重量份,如10、15、20、25、30、35、40重量份等;所述第二发泡剂的含量为1-10重量份,如1、2、3、4、5、6、7、8、9、10重量份等;所述膨胀剂的含量为1-15重量份,如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15重量份等。
[0049]
所述聚氨酯型固化剂具有式(i)所示的结构:
[0050][0051]
式(i)中,r1为c
1-c
10
的亚烷基、c
6-c
20
的亚环烷基或c
6-c
20
的亚芳基,r2为衍生自亲水性低聚物多元醇的基团,r3为带有至少两个巯基的c
1-c
30
的巯基烷基或c
1-c
30
的巯基酯基,n为大于等于1的整数。
[0052]
在一种优选实施方式中,r1为c
4-c8的亚烷基、c
6-c
13
的亚环烷基或c
6-c
13
的亚芳基。其中,c
4-c8的亚烷基的具体实例包括但不限于:亚正丁基、亚异丁基、亚叔丁基、亚正戊基、亚异戊基、亚新戊基、亚正己基、亚异己基、亚叔己基、亚正庚基、亚异庚基、亚叔庚基、亚正辛基、亚异辛基或亚叔辛基,优选为亚正戊基或亚正己基。c
6-c
13
的亚环烷基的具体实例包括但不限于:环己基、降冰片烷二亚甲基、1,1,3-三甲基环己基或甲基二环己基。c
6-c
13
的亚芳基的具体实例包括但不限于:亚苯基、亚甲苯基、亚间二甲基苯基或1,1
’‑
亚甲基二苯基。
[0053]
在一种优选实施方式中,r2具有式(ii)所示的结构:
[0054][0055]
式(ii)中,r
21
为c
1-c
10
的亚烷基,r
22
、r
23
和r
24
各自独立地为c
1-c5的亚烷基或不存在,m为大于等于1的整数。其中,c
1-c
10
的亚烷基的具体实例包括但不限于:亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁基、亚异丁基、亚叔丁基、亚正戊基、亚异戊基、亚新戊基、亚正己基、亚异己基、亚叔己基、亚正庚基、亚异庚基、亚叔庚基、亚正辛基、亚异辛基或亚叔辛基。c
1-c5的亚烷基的具体实例包括但不限于:亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁
基、亚异丁基、亚叔丁基、亚正戊基、亚异戊基或亚新戊基。
[0056]
在一种优选实施方式中,r3具有式(iii)所示的结构:
[0057][0058]
式(iii)中,r
31
为c
1-c5的烷基或-o-co-r
31-sh,r
31
为c
1-c5的亚烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。当r3为三官能度时,r
31
为c
1-c5的烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。从原料易得性的角度考虑,特别优选地,r
31
为甲基,r
32
、r
33
和r
34
各自独立地为亚甲基、亚乙基或亚正丙基。当r3为四官能度时,r
31
为-o-co-r
31-sh,r
31
为c
1-c5的亚烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。从原料易得性的角度考虑,特别优选地,r
31
与r
32
、r
33
和r
34
优选全部相同。
[0059]
在一种具体实施方式中,所述聚氨酯型固化剂按照以下方法制备得到:
[0060]
s1、在惰性气氛下,将式(iv)所示的多异氰酸酯与式(v)所示的亲水性低聚物多元醇进行聚加成反应,得到仅裸露异氰酸酯基团在分子链末端的α,ω-双异氰酸酯聚氨酯前体;
[0061]
s2、在惰性气氛下,将步骤s1中所得α,ω-双异氰酸酯聚氨酯前体与式(ⅵ)所示的含巯基化合物在催化剂的作用下进行聚加成反应,得到聚氨酯型固化剂;
[0062]
ocn-r
1-nco
ꢀꢀ
(式iv)
[0063][0064]
式(iv)中,r1为c
1-c
10
的亚烷基、c
6-c
20
的亚环烷基或c
6-c
20
的亚芳基;式(v)中,r
21
为c
1-c
10
的亚烷基,r
22
、r
23
和r
24
各自独立地为c
1-c5的亚烷基或不存在,m为大于等于1的整数;式(ⅵ)中,r
31
为c
1-c5的烷基或-o-co-r
31-sh,r
31
为c
1-c5的亚烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。
[0065]
所述多异氰酸酯的具体实例包括但不限于:甲苯二异氰酸酯、降冰片烷二异氰酸酯、异佛尔酮二异氰酸酯、间苯二甲基二异氰酸酯、4,4-二环已基加完二异氰酸酯、五亚甲基二异氰酸酯和六亚甲基二异氰酸酯中的至少一种,更优选为五亚甲基二异氰酸酯和/或六亚甲基二异氰酸酯。
[0066]
在一种具体实施方式中,所述亲水性低聚物多元醇通过将式(
ⅷ
)所示的二元醇与
式(
ⅸ
)所示的二元羧酸进行缩合反应得到,其反应过程如式(
ⅹ
)所示;
[0067]
ho-r
21-oh
[0068]
式(viii)
[0069][0070]
式(
ⅷ
)和式(
ⅹ
)中,r
21
为c
1-c
10
的亚烷基;式(
ⅸ
)和式(
ⅹ
)中,r
22
、r
23
和r
24
各自独立地为c
1-c5的亚烷基或不存在。
[0071]
所述二元醇的具体实例包括但不限于:乙二醇、1,2-丙二醇、1,3-丙二醇、2-甲基-1,3-丙二醇、1,4-丁二醇、1,5-戊二醇、新戊二醇、一缩二乙二醇、3-甲基-1,5-戊二醇和1,6-己二醇中的至少一种。所述二元羧酸特别优选为酒石酸。所述二元醇与二元羧酸的摩尔比优选为1:(1.1-1.3)。此外,所述缩合反应通常在催化剂的存在下进行,所述催化剂可以选自硫酸氢钠、对甲苯磺酸、浓硫酸和浓盐酸中的至少一种。
[0072]
在一种更具体的实施方式中,所述亲水性低聚物多元醇通过以下方法制备得到:在惰性气体保护下,将等摩尔量的二元醇和二元羧酸加入反应器中,加入单体总重量0.2-1%的催化剂,将温度控制在60-80℃下搅拌1-5h进行脱水缩聚反应,在升温过程中控制冷凝管的顶部温度低于100℃,期间每间隔0.5h对反应体系抽真空约5min,以除去小分子副产物,使得反应平衡直至反应体系重量无明显变化后(抽出的小分子质量基本不再增加),加入单体总重量0.2-1%的催化剂,继续滴加与二元羧酸的摩尔比为1:(0.1-0.3)的二元醇,之后搅拌反应0.5-2h,以确保合成物质的两端为羟基,最后将温度升高至130-180℃、真空度≥0.095mpa,继续反应0.5-1h,测试羟值达到50-136mgkoh/g,得到数均分子量为500-3000的亲水性低聚物二元醇。
[0073]
所述含巯基化合物具有式(ⅵ)所示的结构。其中,当含巯基化合物为三官能度时,r
31
为c
1-c5的烷基,优选为甲基;r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基,优选各自独立地为亚甲基、亚乙基或亚正丙基。从原料易得性的角度出发,当含巯基化合物为三官能度时,其特别优选选自三羟甲基丙烷三(2-巯基乙酸酯)、三羟甲基丙烷三(3-巯基丙酸酯)和三羟甲基丙烷三(3-巯基丁酸酯)中的至少一种。当硫醇单体为四官能度时,r
31
为-o-co-r
31-sh,r
31
为c
1-c5的亚烷基,r
32
、r
33
和r
34
各自独立地为c
1-c5的亚烷基。从原料易得性的角度出发,当含巯基化合物为四官能度时,其特别优选选自季戊四醇四(巯基乙酸酯)、四(3-巯基丙酸)季戊四醇酯和四(3-巯基丁酸)季戊四醇酯中的至少一种。
[0074]
由于异氰酸酯对水分敏感,因此为了使多异氰酸酯与亲水性低聚物多元醇、含巯基化合物的聚加成反应能够顺利进行,在反应加料前需要对反应容器和反应原料进行除水处理并于将聚加成反应置于惰性气氛下进行。其中,对反应容器进行除水处理的方式通常可以对反应容器进行干燥,具体可以在130-150℃条件下对反应容器干燥2-4h。对反应原料进行除水处理的方式通常可以将反应原料采用冷冻干燥、分子筛除水、手套箱抽换气等方
式进行处理。将聚加成反应置于惰性气氛下进行的方式通常可以往反应容器中通入化学惰性气体、抽换真空等方式以驱除空气并充入惰性气体,从而使反应体系能够保持在惰性气氛下,其中,所述化学惰性气体具体可以为氮气或氩气,优选为氮气。
[0075]
本发明对聚加成反应的条件没有特别的限定,但为了控制反应速率同时兼具聚合物的分子量,步骤s1中涉及的聚加成反应条件通常包括反应温度可以为90-120℃,反应时间可以为2-5h。步骤s2中涉及的聚加成反应条件通常包括反应温度可以为20-50℃,反应时间可以为2-10h。需要说明的是,本发明中聚氨酯型固化剂的制备优选采用“一锅法”的方式进行,即在合成α,ω-双异氰酸酯聚氨酯前体后,直接于上述反应体系中继续添加含巯基化合物和催化剂,而无需进一步处理。此外,本发明中所述聚加成反应通常在有机溶剂的存在下进行,所述有机溶剂为不含有活泼氢的化合物,优选选自甲苯、二甲苯、丙酮、甲乙酮、环己酮、四氢呋喃、二氧六环和二甲基甲酰胺中的至少一种。
[0076]
本发明中,为了得到仅裸露异氰酸酯基团在分子链末端的α,ω-双异氰酸酯聚氨酯前体,所述多异氰酸酯的摩尔量需要比亲水性低聚物多元醇的摩尔量稍过量,优选两者的摩尔比为1:(0.6-0.8),如1:0.6、1:0.65、1:7、1:7.5、1:0.8等。进一步地,所述多异氰酸酯与含巯基化合物的摩尔比优选为1:(0.2-1),如1:0.2、1:0.3、1:0.4、1:0.5、1:0.6、1:0.7、1:1:0.8、1:0.9、1:1等。
[0077]
为了使步骤s1中所得α,ω-双异氰酸酯聚氨酯前体分子链末端的异氰酸酯基团反应完全,步骤s2中通过测试巯基含量的方法来检验反应程度,当巯基的含量达到5-20%时停止反应。由于反应过程会消耗巯基,每隔2小时测试一次巯基含量,直至巯基含量不再降低,即说明反应不再进行,最终的巯基含量为5-20%。所述巯基含量的测定根据cn201711190623.x中提供的测试方法对其进行测试,即利用乙腈溶剂溶解样品后,加入含有与样品相同质量的碘-乙醇溶液、含有两倍样品质量的ki水溶液与巯基发生反应,最后用硫代硫酸钠标准溶液测定剩余碘,从而测得巯基的含量。
[0078]
由于亲水性低聚物多元醇中羟基的活性大,其与异氰酸酯可以直接反应,因此,步骤s1中的聚加成反应不需要额外添加催化剂就能顺利反应。步骤s2中的聚加成反应常用的催化剂为有机叔胺类和/或金属有机化合物类,优选为金属有机化合物类。其中,所述金属有机化合物类的具体实例包括但不限于:异酸亚锡、氯化三甲基锡、二月桂酸二丁基锡、二氯化二丁基锡和三氯化甲基锡中的至少一种。此外,本发明对所述催化剂的用量没有特别地限制,但为了兼顾控制催化速率与聚氨酯型固化剂分子量的大小,所述催化剂的用量优选为单体总重量的0.1wt%-0.3wt%,具体地,可以为0.1wt%、0.15wt%、0.2wt%、0.25wt%、0.3wt%。所述单体总质量为多异氰酸酯、亲水性低聚物多元醇和含巯基化合物的总质量。
[0079]
此外,所述聚氨酯型固化剂在25℃下的粘度优选为3000-30000cps。
[0080]
所述环氧树脂可以为具有两个以上环氧基的脂肪族环氧树脂,也可以为具有两个以上环氧基的芳香族环氧树脂,还可以为两者的混合物,优选为双酚a环氧树脂和/或双酚f环氧树脂。
[0081]
为了便于描述,将组分a中所含的填料称为“第一填料”,将组分b中所含的填料称为“第二填料”。本发明对第一填料和第二填料的种类没有特别的限定,可以为本领域的常规选择,例如,可以各自独立地选自滑石粉、钛白粉、高岭土、碳酸钙、硫酸钡和氧化锌中的
至少一种。
[0082]
为了便于描述,将组分a中所含的发泡剂称为“第一发泡剂”,将组分b中所含的发泡剂称为“第二发泡剂”。本发明对第一发泡剂和第二发泡剂的种类没有特别的限定,可以为本领域的常规选择,例如,可以各自独立地选自碳酸氢钙、碳酸铵、偶氮二异丁腈、偶氮二甲酸二乙酯、对苯磺酰肼以及4,4
’‑
二磺酰肼二苯醚中的至少一种。
[0083]
本发明对防沉降剂的种类没有特别的限定,优选为有机膨润土。
[0084]
所述聚氨酯型固化剂中存在硫醇基团(不存在叔胺),所述硫醇基团会与含叔胺催化剂中的叔胺反应产生硫醇离子,之后该硫醇离子再和环氧基团进行交联固化反应。所述含叔胺催化剂的使用能够降低以上反应所需的活化能。本发明对含叔胺催化剂的种类没有特别的限定,优选为改性脂肪叔胺。其中,所述改性脂肪叔胺所采用的改性方式可以为桐油改性和/或葵二酸改性,所述改性脂肪叔胺的具体实例包括但不限于:yth-201、ty-300、ty-650、g-720、g-725、l-220p,l-2505,l-2801、up-0639、up-583中的至少一种。
[0085]
所述膨胀剂所起的作用为高温水煮过程帮助固化后胶体膨胀增加与水的接触面积。本发明对膨胀剂的种类没有特别的限定,优选为钙矾石。
[0086]
本发明提供的双组分环氧胶黏剂的制备方法包括以下步骤:(1)将环氧树脂、第一填料、第一发泡剂和防沉降剂按比例混合均匀,抽真空除泡后,得到组分a;(2)将聚氨酯型固化剂、含叔胺催化剂、第二填料、第二发泡剂和膨胀剂按比例混合均匀,抽真空除泡后,得到组分b;(3)将组分a和组分b混合均匀,得到双组分环氧胶黏剂。其中,三个步骤的混合均可以在双行星反应釜中进行。
[0087]
以下将通过实施例对本发明进行详细描述。
[0088]
制备例1该制备例用于说明亲水性低聚物多元醇的制备
[0089]
将装有磁力搅拌器、温度计、n2进口管和空气-冷却冷凝管的四口烧瓶安装在覆套式电阻加热器中,加入120.1g酒石酸、83.3g新戊二醇和1g浓硫酸,在氮气保护下搅拌2h,将烧瓶温度控制在70℃进行脱水缩聚反应,在升温过程中控制冷凝管的顶部温度低于100℃,期间每间隔0.5h对反应体系抽真空约5min,以除去小分子副产物,使得反应平衡直至反应体系重量无明显变化后;之后加入20.8g新戊二醇继续反应1h后,再逐渐加热升温至150℃、真空度≥0.095mpa,继续反应使羟值达到120mg koh/g,得到数均分子量为900的亲水性低聚物多元醇,记为a。
[0090]
制备例2该制备例用于说明聚氨酯型固化剂的制备
[0091]
将180g(0.2mol)亲水性低聚物多元醇a加入装有搅拌三口烧瓶中,加热至120℃抽真空脱水1h,降温至90℃,分2次逐步添加50.4g(0.3mol)六亚甲基二异氰酸酯,在惰性气体氛围下搅拌2h;然后加入79.7g(0.2mol)三羟甲基丙烷三(3-巯基丙酸酯)和0.35g(0.11wt%)二丁基二氯化锡催化剂,在温度40℃下反应3.5h,检测巯基含量在15%左右时,终止反应,得到粘稠状液体,该粘稠状液体即为聚氨酯型固化剂,其25℃粘度为5000cps,记为a-1。
[0092]
制备例3该制备例用于说明聚氨酯型固化剂的制备
[0093]
将216g(0.24mol)亲水性低聚物多元醇a加入装有搅拌三口烧瓶中,加热至130℃抽真空脱水0.6h,降温至110℃,加入78.6g(0.3mol)二环已基甲烷-4,4-二异氰酸酯,在惰性气体氛围下搅拌3h;然后加入73.4g(0.20mol)四(3-巯基丙酸)季戊四醇酯和0.55g
(0.15wt%)二丁基二氯化锡催化剂,在温度50℃下反应4h,检测巯基含量在17%左右时,终止反应,得到粘稠状液体,该粘稠状液体即为聚氨酯型固化剂,其25℃粘度为15000cps,记为a-2。
[0094]
制备例4该制备例用于说明聚氨酯型固化剂的制备
[0095]
将216g(0.24mol)亲水性低聚物多元醇a加入装有搅拌三口烧瓶中,加热至120℃抽真空脱水1h,降温至100℃,加入71.14g(0.32mol)异佛尔酮二异氰酸酯,在惰性气体氛围下搅拌3.5h;然后加入79.7g(0.2mol)三羟甲基丙烷三(3-巯基丙酸酯)和0.92g(0.25wt%)二月桂酸二丁基锡催化剂,在温度45℃下反应4h,检测巯基含量在13%左右时,终止反应,得到粘稠状液体,该粘稠状液体即为聚氨酯型固化剂,其25℃粘度为9000cps,记为a-3。
[0096]
对比制备例1该对比制备例用于说明聚脲固化剂的制备
[0097]
在1l的双行星混合动力反应釜中加入201.6g(1.20mol)六亚甲基二异氰酸酯,然后称取48.8g(0.82mol)乙二胺,分2次逐步添加乙二胺溶液到反应釜中,在温度为5℃下反应3h;然后加入318.6g(0.80mol)三羟甲基丙烷三(3-巯基乙酸酯)和0.6g(0.11wt%)二丁基二氯化锡催化剂,在温度为25℃下反应4h,检测巯基含量在15%左右,终止反应,得到粘稠状液体,该粘稠状液体即为聚脲固化剂,其25℃粘度为7000cps,记为da-1。
[0098]
实施例1该实施例用于说明双组分环氧胶黏剂的制备
[0099]
(1)原料:
[0100]
a组分按重量份的制备原料如下:
[0101][0102]
b组分按重量份的制备原料如下:
[0103][0104]
(2)制备过程:
[0105]
将环氧树脂、填料、发泡剂、防沉降剂按比例加入双行星反应釜中,搅拌2小时,抽真空1h除泡后,即得组分a;
[0106]
将聚氨酯型固化剂a-1、改性胺、填料、发泡剂、膨胀剂按比例加入双行星反应釜中,搅拌2小时,抽真空1h除泡后,即得组分b;
[0107]
将组分a和组分b按质量比1:1的比例混合均匀即可得双组分环氧胶黏剂,记为d1。
[0108]
实施例2该实施例用于说明双组分环氧胶黏剂的制备
[0109]
(1)原料:
[0110]
a组分按重量份的制备原料如下:
[0111][0112]
b组分按重量份的制备原料如下:
[0113][0114]
(2)制备过程:
[0115]
将环氧树脂、填料、发泡剂、防沉降剂按比例加入双行星反应釜中,搅拌2小时,抽真空1h除泡后,即得组分a;
[0116]
将将聚氨酯型固化剂a-2、改性胺、填料、发泡剂、膨胀剂按比例加入双行星反应釜中,搅拌2小时,抽真空1h除泡后,即得到组分b;
[0117]
将组分a和组分b按质量比1:1的比例混合均匀即可得双组分环氧胶黏剂,记为d2。
[0118]
实施例3该实施例用于说明双组分环氧胶黏剂的制备
[0119]
(1)原料:
[0120]
a组分按重量份的制备原料如下:
[0121][0122]
b组分按重量份的制备原料如下:
[0123][0124]
(2)制备过程:
[0125]
将环氧树脂、填料、发泡剂、防沉降剂按比例加入双行星反应釜中,搅拌2小时,抽
真空1h除泡后,即得到组分a;
[0126]
将聚氨酯型固化剂a-3、改性胺、填料、发泡剂、膨胀剂按比例加入双行星反应釜中,搅拌2小时,抽真空1h除泡后,即得到组分b;
[0127]
将组分a和组分b按质量比1:1的比例混合均匀即可得双组分环氧胶黏剂,记为d3。
[0128]
对比例1
[0129]
按照实施例1的方法制备双组分环氧胶黏剂,不同的是,将聚氨酯型固化剂a-1采用相同重量份的三羟甲基丙烷三(3-巯基丙酸酯)替代,其余条件与实施例1相同,得到参比双组分环氧胶黏剂,记为da1。
[0130]
对比例2
[0131]
按照实施例2的方法制备双组分环氧胶黏剂,不同的是,将聚氨酯型固化剂a-2采用相同重量份的四(3-巯基丙酸)季戊四醇酯替代,其余条件与实施例2相同,得到参比双组分环氧胶黏剂,记为da2。
[0132]
对比例3
[0133]
按照实施例3的方法制备双组分环氧胶黏剂,不同的是,将聚氨酯型固化剂a-3采用相同重量份的三羟甲基丙烷三(3-巯基丙酸酯)替代,其余条件与实施例3相同,得到参比双组分环氧胶黏剂,记为da3。
[0134]
对比例4
[0135]
按照实施例1的方法制备双组分环氧胶黏剂,不同的是,将聚氨酯型固化剂a-1采用相同重量份的聚脲固化剂da-1替代,其余条件与实施例1相同,得到参比双组分环氧胶黏剂,记为da4。
[0136]
对比例5
[0137]
按照实施例1的方法制备双组分环氧胶黏剂,不同的是,将组分a中的对苯磺酰肼采用相同重量份的碳酸钙替代,同时将组分b中的碳酸铵采用相同重量份的碳酸钙替代,其余条件与实施例1相同,得到参比双组分环氧胶黏剂,记为da5。
[0138]
对比例6
[0139]
按照实施例1的方法制备双组分环氧胶黏剂,不同的是,将组分b中的钙矾石采用相同重量份的碳酸铵替代,其余条件与实施例1相同,得到参比双组分环氧胶黏剂,记为da6。
[0140]
测试例
[0141]
(1)剪切强度和拉伸强度:按照gb7124-86《胶黏剂拉伸剪切强度测定方法(金属-金属)》对各实施例和对比例所得胶黏剂的剪切强度和拉伸强度进行测试,其中,粘接形式采用不锈钢粘接不锈钢,常温固化24h。所得结果见表1。
[0142]
(2)吸水率:将各实施例和对比例所得胶黏剂在常温下固化24h,将固化后的胶黏剂制成60mm
×
60mm
×
1mm的方形试样,之后根据gb/t 1034-2008《塑料吸水性的测定》测试其在23℃水中浸泡24小时后的吸水量,记录吸水质量分数(%)。所得结果见表1。
[0143]
(3)水煮脱落时间:将以上各实施例和对比例所得胶黏剂分别涂覆在钢板上,用钢化玻璃板搭接压合,制作试验样品,粘接面积为25.4mm
×
5mm,并保证胶层的厚度为0.1mm,对试验样品分别进行固化(室温,24h),然后将固化完全的样品放入100℃的热水中,计算从放入热水中至钢板和玻璃板自动分离的时间。所得结果见表1。
[0144]
表1
[0145][0146]
从以上结果可以看出,本发明提供的双组分环氧胶黏剂在水煮环境下可以快速脱胶,提高生产效率,节约能源,将其作为光伏切割用胶黏剂时能够提高清胶效率。从实施例1-3与对比例1-6的对比可以看出,本发明提供的双组分环氧胶黏剂中,所述聚氨酯型化合物与发泡剂和膨胀剂能够起到良好的协同配合作用,以使得胶层在水煮环境中能够快速脱落。
[0147]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在不脱离本发明的原理和宗旨的情况下在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。