蛋白质-抗病毒化合物偶联物
政府许可权
1.本发明是根据美国卫生与公众服务部授予的hhso100201700020c协议在政府支持下完成。该政府对本发明享有一定的权利。相关申请的交叉引用
2.本技术根据35u.s.c.
§
119要求并享有于2020年1月24日提交的美国临时申请号62/965,735以及于2020年10月20日提交的美国临时申请号63/094,285的权益,其内容均通过引用其全文并入本发明。
发明领域
3.本发明提供了抗病毒化合物及其蛋白质偶联物,以及用于治疗多种疾病、病症和病况的方法,包括施用所述抗病毒化合物及其蛋白质偶联物。发明背景
4.流感是高度传染性疾病,其历史悠久,以大流行、流行病、死灰复燃和暴发为特征。尽管每年皆在进行疫苗接种工作,但流感感染仍会导致大量发病率和死亡率。
5.流感病毒由甲、乙和丙三种主要类型组成。基于编码表面糖蛋白(血凝素(ha)和神经氨酸酶(na),其均是病毒附着以及进入宿主细胞所必需的)的两个基因的抗原区域中的等位基因变异,可将甲型流感病毒分成亚型。
6.血凝素是一种三聚体糖蛋白,其包含两个结构域,即,由受体结合位点组成的球状头部结构域(其经常发生抗原漂移)和茎区(其在各种流感病毒株中更为保守)。ha蛋白被合成为前体(ha0),经过蛋白水解加工产生两个亚基(ha1和ha2),此两个亚基彼此之间相互结合以形成茎/球状头部结构。ha1肽负责将病毒附着至细胞表面。ha2肽形成茎状结构,其可介导内体中病毒与细胞膜的融合,从而使核糖核蛋白复合物释放进入细胞质。
7.目前,有18种亚型由其血凝素蛋白(h1-h18)定义。18种ha可分为两组。组1由h1、h2、h5、h6、h8、h9、h11、h12、h13、h16、h17和h18亚型组成,组2包括h3、h4、h7、h10、h14和h15亚型。
8.尽管进行了数十载的研究,但还并未出现可广泛中和或抑制甲型流感病毒感染或减弱由甲型流感病毒引起的疾病的市售抗体或抗体-药物偶联物(adc)。因此,需要识别能够中和多种甲型流感病毒亚型的新抗体和adc,并可用作预防或治疗甲型流感感染的药物。
发明摘要
9.本发明提供了可用于例如抗病毒治疗的化合物。在某些实施方案,所述化合物包括vx-787及其衍生物、巴洛沙韦(baloxavir)及其衍生物、和/或巴洛沙韦酯(baloxavir marboxil)及其衍生物。在一个实施方案,本发明提供了抗体-药物偶联物,其包括与有效负载(例如,抗病毒化合物)、连接体-有效负载(例如,连接体-抗病毒化合物)、和/或本发明所述化合物偶联的抗流感抗体或其抗原结合片段。
10.在某些实施方案,本发明提供了具有以下结构的化合物:
其中l是连接体;ba是结合剂;和k是从1至30的整数。
11.在某些实施方案,本发明提供了具有以下结构的连接体-有效负载(例如,连接体-抗病毒化合物):或其药学上可接受的盐,其中l是连接体;和rg是活性基团部分。
12.在另一实施方案,本发明阐述了制备本发明所述有效负载或化合物、连接体-有效负载或抗体-药物偶联物及组合物的方法。
13.在另一实施方案,本发明提供了治疗、预防、减少或抑制受试者的如本发明所述的与感染相关的疾病、病症或病况的方法,其包括向所述受试者施用有效量的如本发明所述的有效负载(例如,抗病毒化合物)、连接体-有效负载(例如,连接体-抗病毒化合物)、抗体-药物偶联物、或药物组合物。附图简要说明
14.图1a示出了通过lc-ms测定的11729-q295-11的强度加权平均连接体-有效负载(例如,连接体-抗病毒化合物)载量。图1b示出了通过lc-ms测定的同型对照-q295-11的强度加权平均连接体-有效负载(例如,连接体-抗病毒化合物)载量。
15.图2a示出了通过lc-ms测定的11729-hc-c端-11的强度加权平均连接体-有效负载(例如,连接体-抗病毒化合物)载量。图2b示出了通过lc-ms测定的同型对照-hc-c端-11的强度加权平均连接体-有效负载(例如,连接体-抗病毒化合物)载量。
16.图3示出了11729-hc-c端-11、11729-hc-n端-11、11729-lc-c端-11和11729-lc-n端-11与甲型流感感染细胞的elisa亚纳摩尔特异性结合。
17.图4示出了11729-hc-c端-11、11729-lc-c端-11、11729-lc-n端-11、mab11729以及同型对照抗体和同型对照-hc-c端-11对抗甲型流感感染的抗病毒功效比较。
18.图5示出了72小时后11729-hc-c端-11在人血浆和猴血浆中的体外稳定性(即,没有连接体-有效负载(例如连接体-抗病毒化合物)损失;或没有dar降低)。
19.图6示出了11729-hc-c端-11以及同型对照抗体和同型对照-hc-c端-11对抗甲型流感感染的抗病毒功效比较。
20.图7示出了11729-hc-c端-6、11729-lc-c端-6、mab11729以及同型对照抗体、同型对照-hc-c端-6抗体和同型对照-lc-c端-6抗体对抗甲型流感感染的抗病毒功效比较。
21.图8示出了11729-hc-c端-45a、11729-lc-c端-45a、11729-hc-c端-34、11729-lc-c端-34、mab11729以及同型对照抗体和同型对照-抗体-药物偶联物对抗甲型流感感染的抗病毒功效比较。
22.图9示出了5385-hc-c端-11、5385-lc-c端-11、mab11729以及同型对照抗体和同型对照-抗体-药物偶联物对抗甲型流感感染的抗病毒功效比较。
23.图10示出了11729-hc-c端-45d、mab11729以及同型对照抗体和同型对照-hc-c端-45d对抗甲型流感感染的抗病毒功效比较。
24.图11示出了11729-hc-c端-45d与甲型流感感染细胞的特异性结合。
具体实施方式
25.本发明提供了可用于治疗例如受试者流感感染的化合物、组合物及方法。定义
26.当提及本发明提供的化合物时,除非另有说明,否则以下术语具有下列含义。除非另有定义,否则本发明使用的所有技术术语和科学术语均具有与本领域普通技术人员通常所理解的相同的含义。在本发明提供的术语存在多个定义的情况下,除非另有说明,否则以所述这些定义为准。
27.短语“流感血凝素”,亦称为“流感ha”,是在流感病毒体表面上发现的三聚体糖蛋白,其介导病毒附着(经由ha1结合至a-2,3-唾液酸和a-2,6-唾液酸)以及进入(通过构象变化)宿主细胞。ha由两个结构域组成:包含受体结合位点的球状头部结构域(其受到高频率的抗原突变)和茎区(其在各种流感病毒株中更为保守)。流感ha被合成为前体(ha0),经过蛋白水解加工产生两个亚基(ha1和ha2),此两个亚基彼此之间相互结合以形成茎/球状头部结构。病毒ha是所述病毒上变异最多的抗原(18种亚型可分成两组),但茎(ha2)在每组内高度保守。
28.全长流感ha的氨基酸序列以genbank中登录号为fj966082.1提供的流感分离株h1 n1 a/california/04/2009的氨基酸序列为例。短语“流感-ha”还包括分离自不同流感分离株(例如,gq149237.1、nc_002017、km972981.1等)的流感ha蛋白变体。短语“流感-ha”亦包括重组流感ha或其片段。所述短语亦包含同例如组氨酸标签、小鼠或人fc、或信号序列偶联的流感ha或其片段。
29.本发明使用的短语“流感感染”亦表征为“流感”,是指由流感病毒引起的严重急性呼吸道疾病。所述短语包括呼吸道感染及症状,包括高烧、头痛、全身酸痛、疲劳和虚弱,在一些案例中,极度疲惫、鼻塞、打喷嚏、喉咙痛、胸部不适、咳嗽、呼吸急促、支气管炎、肺炎,严重者死亡。
30.本发明使用的“烷基”是指一价且饱和的烃基基团部分。烷基任选地被取代,且可以是直链、支链或环状的(即环烷基)。烷基包括但不限于具有1-20个碳原子,即c
1-20
烷基;1-12个碳原子,即c
1-12
烷基;1-8个碳原子,即c
1-8
烷基;1-6个碳原子,即c
1-6
烷基;和1-3个碳原子,即c
1-3
烷基的那些基团。烷基基团部分的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、异丁基、戊基基团部分、己基基团部分、环丙基、环丁基、环戊基
和环己基。戊基基团部分包括但不限于正戊基和异戊基。己基基团部分包括但不限于正己基。
31.本发明使用的“亚烷基”是指二价烷基基团。除非另有说明,否则亚烷基包括但不限于1-20个碳原子。亚烷基基团如本发明针对烷基所描述的那样任选地被取代。在一些实施方案,亚烷基是未取代的。
32.在未指定其立体化学的情况下,氨基酸或氨基酸残基的命名旨在涵盖氨基酸的l形式、氨基酸的d形式或其外消旋混合物。
33.本发明使用的“卤代烷基”是指如上所定义的烷基,其中所述烷基包括至少一个选自卤素例如氟(f)、氯(cl)、溴(br)或碘(i)的取代基。卤代烷基的实例包括但不限于,
–
cf3、
–
ch2cf3、
–
ccl2f和
–
ccl3。
34.本发明使用的“烯基”是指包含至少两个碳原子和一个或多个非芳族碳-碳双键的一价烃基基团部分。烯基任选地被取代,且可以是直链、支链或环状的。烯基包括但不限于具有2-20个碳原子,即c
2-20
烯基;2-12个碳原子,即c
2-12
烯基;2-8个碳原子,即c
2-8
烯基;2-6个碳原子,即c
2-6
烯基;和2-4个碳原子,即c
2-4
烯基的那些基团。烯基基团部分的实例包括但不限于乙烯基、丙烯基、丁烯基和环己烯基。
35.本发明使用的“炔基”是指包含至少两个碳原子和一个或多个碳-碳三键的一价烃基基团部分。炔基任选地被取代,且可以是直链、支链或环状的。炔基包括但不限于具有2-20个碳原子,即c
2-20
炔基;2-12个碳原子,即c
2-12
炔基;2-8个碳原子,即c
2-8
炔基;2-6个碳原子,即c
2-6
炔基;和2-4个碳原子,即c
2-4
炔基的那些基团。炔基基团部分的实例包括但不限于乙炔基、丙炔基和丁炔基。
36.本发明使用的“烷氧基”是指一价且饱和的烃基基团部分,其中所述烃包括与氧原子连接的单键,和其中所述自由基位于氧原子上,例如乙氧基ch3ch2–o·
。烷氧基取代基通过所述烷氧基取代基的氧原子与它们取代的化合物连接。烷氧基任选地被取代,且可以是直链、支链或环状的,即环烷氧基。烷氧基包括但不限于具有1-20个碳原子,即c
1-20
烷氧基;1-12个碳原子,即c
1-12
烷氧基;1-8个碳原子,即c
1-8
烷氧基;1-6个碳原子,即c
1-6
烷氧基;和1-3个碳原子,即c
1-3
烷氧基的那些。烷氧基基团部分的实例包括但不限于甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、异丁氧基、戊氧基基团部分、己氧基基团部分、环丙氧基、环丁氧基、环戊氧基和环己氧基。
37.本发明使用的“卤代烷氧基”是指如上所定义的烷氧基,其中所述烷氧基包括至少一个选自卤素例如f、cl、br或i的取代基。
38.本发明使用的“芳基”是指一价基团部分,其是芳族化合物的原子团,其中所述环原子均为碳原子。芳基任选地被取代,且可以是单环或多环的,例如双环或三环。芳基基团部分的实例包括但不限于具有6至20个环碳原子,即c
6-20
芳基;6至15个环碳原子,即c
6-15
芳基,和6至10个环碳原子,即c
6-10
芳基的那些。芳基基团部分的实例包括但不限于苯基、萘基、芴基、薁基、蒽基、菲基和芘基。
39.本发明使用的“芳基烷基”或“芳烷基”是指烷基化合物原子团的一价基团部分,其中所述烷基化合物被芳族取代基取代,即,所述芳族化合物包括与烷基基团连接的单键,和其中所述自由基位于所述烷基基团上。芳烷基基团通过所述烷基基团与所示的化学结构连
接。芳烷基可由以下结构表示,例如,接。芳烷基可由以下结构表示,例如,其中b是芳族基团部分,例如芳基或苯基。芳烷基任选地被取代,即所述芳基基团和/或所述烷基基团,可以如本发明所述的那样被取代。芳烷基的实例包括但不限于苄基。
40.本发明使用的“烷基芳基”是指芳基化合物原子团的一价基团部分,其中所述芳基化合物被烷基取代基取代,即,所述芳基化合物包括与烷基基团连接的单键,其中所述自由基位于所述芳基基团上。烷基芳基基团通过所述芳基基团与所示的化学结构连接。烷基芳基可由以下结构表示,例如,基可由以下结构表示,例如,其中b是芳族基团部分,例如苯基。烷基芳基任选地被取代,即所述芳基基团和/或所述烷基基团,可以如本发明所述的那样被取代。烷基芳基的实例包括但不限于甲苯甲酰基。
41.本发明使用的“芳氧基/芳基氧基”是指芳族化合物原子团的一价基团部分,其中所述环原子均为碳原子,和其中所述环被氧基取代,即所述芳族化合物包括与氧原子连接的单键,和其中所述自由基位于氧原子上,例如苯氧基芳氧基取代基通过此氧原子与它们取代的化合物连接。芳氧基任选地被取代。芳氧基包括但不限于具有6至20个环碳原子,即c
6-20
芳氧基;6至15个环碳原子,即c
6-15
芳氧基,和6至10个环碳原子,即c
6-10
芳氧基的那些基团。芳氧基基团部分的实例包括但不限于苯氧基、萘氧基和蒽氧基。
42.本发明使用的“亚芳基”是指芳族化合物的二价基团部分,其中所述环原子仅为碳原子。亚芳基任选地被取代,且可以是单环或多环的,例如双环或三环。亚芳基基团部分的实例包括但不限于具有6至20个环碳原子,即c
6-20
亚芳基;6至15个环碳原子,即c
6-15
亚芳基;和6至10个环碳原子,即c
6-10
亚芳基的那些。
43.本发明使用的“杂烷基”是指其中一个或多个碳原子被杂原子替换的烷基。本发明使用的“杂烯基”是指其中一个或多个碳原子被杂原子替换的烯基。本发明使用的“杂炔基”是指其中一个或多个碳原子被杂原子替换的炔基。合适的杂原子包括但不限于氮、氧和硫原子。杂烷基、杂烯基和杂炔基均任选地被取代。杂烷基基团部分的实例包括但不限于氨基烷基、磺酰基烷基、亚磺酰基烷基。杂烷基基团部分的实例还包括但不限于甲氨基、甲磺酰基和甲基亚磺酰基。
44.本发明使用的“杂芳基”是指芳族化合物原子团的一价基团部分,其中所述环原子包含碳原子和至少一个氧、硫、氮或磷原子。杂芳基基团部分的实例包括但不限于具有5至20个环原子,5至15个环原子,和5至10个环原子的那些。杂芳基任选地被取代。
45.本发明使用的“亚杂芳基”是指其中所述芳环的一个或多个环原子被氧、硫、氮或磷原子替换的二价杂芳基。亚杂芳基任选地被取代。
46.本发明使用的“杂环烷基”是指其中一个或多个碳原子被杂原子替换的环烷基。合适的杂原子包括但不限于氮、氧和硫原子。杂环烷基任选地被取代。杂环烷基基团部分的实例包括但不限于吗啉基、哌啶基、四氢吡喃基、吡咯烷基、咪唑烷基、噁唑烷基、噻唑烷基、二氧杂环戊烷基、二硫杂环戊烷基、四氢吡喃基(oxanyl)或四氢噻喃基(thianyl)。
47.本发明使用的“路易斯酸”是指接受孤对电子的分子或离子。本发明所述方法中使
1-基氧基)三吡咯烷基鏻六氟磷酸盐(pybop)、(7-偶氮苯并三氮唑-1-基氧基)三吡咯烷基鏻六氟磷酸盐(pyaop)、三吡咯烷基溴化鏻六氟磷酸盐(pybrop)、o-(苯并三氮唑-1-基)-n,n,n’,n
’‑
四甲基脲六氟磷酸盐(hbtu)、o-(苯并三氮唑-1-基)-n,n,n’,n
’‑
四甲基脲六氟硼酸盐(tbtu)、1-[双(二甲氨基)亚甲基]-1h-1,2,3-三氮唑并[4,5-b]吡啶鎓3-氧化六氟磷酸盐(hatu)、n-乙氧羰基-2-乙氧基-1,2-二氢喹啉(eedq)、n-乙基-n
’‑
(3-二甲氨基丙基)碳二亚胺(edc)、2-氯-1,3-二甲基咪唑鎓六氟磷酸盐(cip)、2-氯-4,6-二甲氧基-1,3,5-三嗪(cdmt)、羰二咪唑(cdi)、和1-氰基-2-乙氧基-2-氧代亚乙基氨基氧基)二甲氨基-吗啉基-碳鎓六氟磷酸盐(comu)。在一些实施例,首先将羧酸转化为活化的羧酸酯,然后将活化的羧酸酯用胺处理形成酰胺键。在一些实施方案,将羧酸用试剂处理。所述试剂通过使羧酸去质子来活化羧酸,然后由于去质子化的羧酸亲核进攻到质子化试剂上,从而与去质子化的羧酸形成产物复合物。对于某些羧酸,相较羧酸转化成活化酯之前,所述活化羧酸酯更容易受到胺的亲核进攻。此举使得酰胺键形成。因此,所述羧酸被描述为活化的。示例性试剂包括dcc和dic。
[0053]
本发明使用的术语“残基”是指化合物内化学反应后保留的化学基团部分。例如,术语“氨基酸残基”或“n-烷基氨基酸残基”是指氨基酸或n-烷基氨基酸与合适的偶联配偶体进行酰胺偶联或肽偶联的产物;其中,例如,在氨基酸或n-烷基氨基酸的酰胺偶联或肽偶联之后将水分子排出,从而得到氨基酸残基或n-烷基氨基酸残基掺入其中的产物。
[0054]
本发明使用的“构造异构体(constitutional isomer)”是指具有相同分子式但由于原子排列方式而产生不同化学结构的化合物。示例性构造异构体包括正丙基和异丙基;正丁基、仲丁基和叔丁基;以及正戊基、异戊基和新戊基等。
[0055]
某些基团、分子/基团部分、取代基、和原子被描述为具有与键相交的波浪线,以指示所述基团、分子/基团部分、取代基、原子通过其进行连接的原子。譬如,被丙基基团取代的苯基基团可表示为:具有下述结构:本发明使用的,通过环原子之间的键连接至环状基团(例如,芳环、杂芳环、稠环、和饱和或不饱和环烷基或杂环烷基)的取代基的例示说明意在表示,除非另有说明,环状基团可根据本发明所阐述的技术或即时公开的涉及本领域已知的技术,在环状基团的任何环位置或稠环基团的任何环上被取代基取代。譬如,所述基团其中下标q是从0至4的整数,和其中取代基r1的位置一般性地描述,即不直接连接至键合线结构的任何顶点,即特定的环碳原子,包括以下,其中取代基r1与特
定环碳原子连接的基团的非限制性实例:定环碳原子连接的基团的非限制性实例:
[0056]
本发明使用的短语“活性(反应性)连接体”或缩写“rl”是指包含活性(反应性)基团(“rg”)和间隔基团(“sp”)的一价基团,如所示,其中rg是所述活性基团,和sp是所述间隔基团。如本发明所述,活性(反应性)连接体可包含一个以上活性(反应性)基团和一个以上间隔基团。所述间隔基团是将活性(反应性)基团桥接至另一个基团例如有效负载(譬如,抗病毒化合物)的任何二价基团部分。所述活性连接体(rl)同与之连接的有效负载(例如,抗病毒化合物)一起,组成可用作制备本发明所述抗体偶联物的合成前体的中间体(“连接体-有效负载”(lp)(例如,连接体-抗病毒化合物))。如本发明所用,有效负载可以是抗病毒化合物,并且掺入所述这些抗病毒化合物的连接体-有效负载可称为“连接体-抗病毒化合物”。所述活性连接体包含活性基团,其是能够与另一基团(例如抗体、经修饰的抗体、或其抗原结合片段)的活性部分反应的官能团或基团部分。由所述活性基团与所述抗体、经修饰的抗体或其抗原结合片段反应产生的基团部分与所述连接基团一起,组成本发明所述的偶联物的“结合剂连接体”(“bl”)部分。在某些实施方案,所述“活性基团”是与抗体或其抗原结合片段的半胱氨酸或赖氨酸残基反应的官能团或基团部分(例如,马来酰亚胺或n-羟基琥珀酰亚胺(nhs)酯)。在一些实施例,所述活性基团是官能团,例如其
与抗体或其抗原结合片段上的半胱氨酸残基反应,与其形成c-s键,例如其中ab是指抗体或其抗原结合片段,s是指半胱氨酸残基上的s原子,所述官能团通过此半胱氨酸残基上的s原子与ab结合。在一些实施例,所述活性基团是官能团,例如其与抗体或其抗原结合片段上的赖氨酸残基反应,与其形成酰胺键,例如其中ab是指抗体或其抗原结合片段,nh是指赖氨酸侧链残基上的nh原子,所述官能团通过此赖氨酸侧链残基上的nh原子与ab结合。
[0057]
本发明使用的短语“可生物降解的基团部分”是指在体内降解为无毒的、可生物相容的组分的基团部分,所述组分可通过普通生物过程从体内清除。在一些实施方案,可生物降解的基团部分在约90天或更短,约60天或更短,或约30天或更短的时间内在体内完全或基本上降解,其中降解的程度基于所述可生物降解基团部分的质量损失百分比,其中完全降解对应于100%质量损失。示例性可生物降解的基团部分包括但不限于脂族聚酯类,例如聚(ε-己内酯)(pcl),聚(3-羟基丁酸酯)(phb),聚(乙醇酸)(pga),聚(乳酸)(pla)和其与乙醇酸的共聚物(即,聚(d,l-丙交酯-共乙交酯)(plga)(vert m,schwach g,engel r and coudane j(1998)j control release 53(1-3):85-92;jain r a(2000)biomaterials 21(23):2475-2490;uhrich k e,cannizzaro s m,langer r s and shakesheff k m(1999)chemical reviews 99(11):3181-3198;和park t g(1995)biomaterials 16(15):1123-1130,各文献均通过引用其全部内容并入本发明)。
[0058]
本发明使用的短语“结合剂连接体”、或“bl”是指使结合剂(例如,抗体或其抗原结合片段)与本发明所述的有效负载化合物(例如,vx-787及其衍生物、和/或巴洛沙韦(baloxavir)及其衍生物(例如巴洛沙韦酯(baloxavir marboxil)及其衍生物))、以及任选地与一个或多个侧链化合物连接、结合或键合的任何二价、三价、或多价基团或基团部分。通常,本发明所述的抗体偶联物的合适结合剂连接体是足以稳定以利用抗体偶联物的循环半衰期,并且同时能够在抗原介导的偶联物内化后释放其有效负载的那些。连接体可以是可切割的或不可切割的。可切割的连接体是内化后由细胞内代谢进行切割的连接体,例如通过水解、还原或酶促反应进行切割。不可切割的连接体是通过内化后抗体的溶酶体降解来释放附接的有效负载的连接体。合适的连接体包括但不限于酸不稳定连接体,水解不稳定连接体,酶促可切割连接体,还原不稳定连接体,自降解(self-immolative)连接体和不可切割连接体。合适的连接体还包括但不限于,是或包含肽类,葡糖苷酸类,琥珀酰亚胺-硫醚类,聚乙二醇(peg)单元类,腙类,马来酰亚胺(mal)-己酰单元类,二肽单元类,缬氨酸-瓜氨酸单元类,和对氨基苄氧基羰基(pabc)单元类、对氨基苄基(pab)单元类的那些。在一些实施方案,所述结合剂连接体(bl)包含由活性连接体(rl)的活性基团(rg)与结合剂(例如,抗体、经修饰的抗体、或其抗原结合片段)的活性部分进行反应形成的基团部分。
[0059]
在一些实施例,所述bl包括以下基团部分:其中是与所述抗体或其抗原结合片段的所述半胱氨酸连接的键。在一些实施例,所述bl包括以下基团部分:其中是与所述抗体或其抗原结合片段的所述赖氨酸连接的键。
[0060]
在一些实施方案,所述结合剂是抗体或其抗原结合片段。所述抗体可以是本领域技术人员已知的任何形式。
[0061]
本发明使用的术语“抗体”是指包含至少一个互补决定区(cdr)的任何抗原结合分子或分子复合物,所述互补决定区(cdr)与特定抗原特异性结合或相互作用。术语“抗体”包括免疫球蛋白分子,所述免疫球蛋白分子包含四条多肽链,通过二硫键相互连接的两条重(h)链和两条轻(l)链(即,完整抗体分子),以及其多聚体(例如igm)或其抗原结合片段。每条重链包含重链可变区(本发明缩写为hcvr或vh)和重链恒定区。所述重链恒定区包含三个结构域,ch1、ch2和ch3。每条轻链包含轻链可变区(本发明缩写为lcvr或v
l
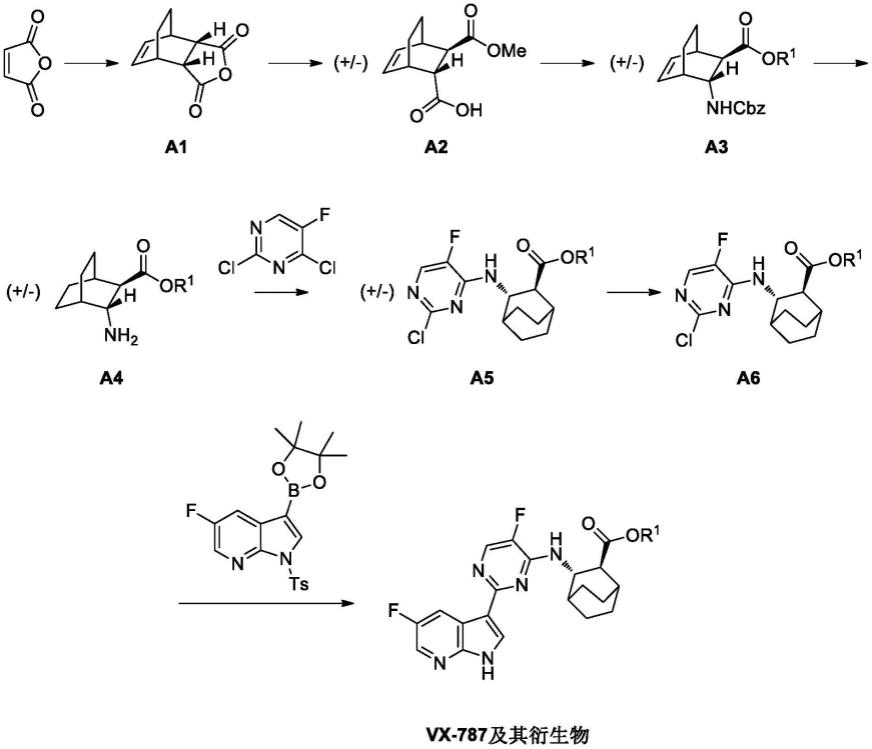
)和轻链恒定区。所述轻链恒定区包含一个结构域(c
l
1)。所述vh区和v
l
区可进一步细分为高变区,称为互补决定区(cdr),散布有更保守的区域,称为框架区(fr)。各vh和v
l
分别由三个cdr和四个fr组成,按照以下顺序从氨基端到羧基端排列:fr1,cdr1,fr2,cdr2,fr3,cdr3,fr4。在本发明公开的不同实施方案中,适用于本发明化合物的所述抗体(或其抗原结合部分)的fr可以与人种系序列相同,或者可以是天然、或人工修饰的。可基于两个或更多个cdr的并列分析来定义氨基酸共有序列。本发明使用的术语“抗体”还包括完整抗体分子的抗原结合片段。本发明使用的术语抗体的“抗原结合结构域”或“抗原结合部分”,抗体的“抗原结合片段”等包括任何天然存在的、可酶促得到的、合成的、或基因工程化的多肽或糖蛋白,其特异性结合抗原以形成复合物。在某些实施方案中,术语“抗原结合片段”是指多特异性抗原结合分子的多肽片段。本发明使用的术语抗体的“抗原结合片段”或“抗体片段”是指保留结合抗原例如流感ha的能力的抗体的一个或多个片段。抗体的抗原结合片段可以例如使用任何合适的标准技术,例如涉及操纵和表达编码抗体可变结构域和任选恒定结构域的dna的蛋白水解消化或重组基因工程技术,由完整抗体分子衍生得到。此种dna是已知的和/或容易从例如商业来源、dna文库(包括例如噬菌体-抗体文库)得到,或可以合成得到。可通过化学方法或通过使用分子生物学技术对dna进行测序和操作,例如,将一个或多个可变结构域和/或恒定结构域排列成合适的构型,或引入密码子,产生半胱氨酸残基,修饰、添加或删除氨基酸等。抗原结合片段的非限制性实例包括:(i)fab片段;(ii)f(ab')2片段;(iii)fd片段;(iv)fv片段;(v)单链fv(scfv)分子;(vi)dab片段;和(vii)由模拟抗体(例如,含cdr的片段、或分离的cdr如cdr3肽)或受约束的fr3-cdr3-fr4肽的高变区的氨基酸残基组成的最小识别单元。其他工程化分子,如结构域特异性抗体类,单结构域抗体类,结构域缺失抗体类,嵌合抗体类,cdr嫁接抗体类,双抗体类,三抗体类,四抗体类,微抗体类,纳米抗体类(例如,一价纳米抗体类,二价纳米抗体类等),小型模块化免疫药物(smip)和鲨鱼可变ignar结构域亦均
包含在本发明所使用的表述“抗原结合片段”内。抗体的抗原结合片段通常包含至少一个可变结构域。所述可变结构域可具有任何尺寸或氨基酸组成,并且通常包含至少一个cdr,所述至少一个cdr与一个或多个框架序列相邻或位于框内。在具有与v
l
结构域相关的vh结构域的抗原结合片段中,所述vh和v
l
结构域可以任何合适的排列相对于彼此进行定位。譬如,所述可变区可以是二聚的并含有v
h-vh、v
h-v
l
或v
l-v
l
二聚体。或者,所述抗体的抗原结合片段可包含单体vh或v
l
结构域。在某些实施方案,抗体的抗原结合片段可包含至少一个可变结构域,所述可变结构域与至少一个恒定结构域共价连接。可在本发明抗体的抗原结合片段内发现的可变结构域和恒定结构域的非限制性示例性构型包括:(i)v
h-ch1;(ii)v
h-ch2;(iii)v
h-ch3;(iv)v
h-ch1-ch2;(v)v
h-ch1-ch2-ch3;(vi)v
h-ch2-ch3;(vii)v
h-c
l
;(viii)v
l-ch1;(ix)v
l-ch2;(x)v
l-ch3;(xi)v
l-ch1-ch2;(xii)v
l-ch1-ch2-ch3;(xiii)v
l-ch2-ch3;和(xiv)v
l-c
l
。在可变结构域和恒定结构域的任何构型中,包括上文列出的任何示例性构型,所述可变结构域和恒定结构域可以彼此直接连接或可以通过完整或部分铰链或连接体区域进行连接。铰链区可由至少2个(例如,5、10、15、20、40、60个或更多个)氨基酸组成,所述氨基酸使得单个多肽分子中相邻可变结构域和/或恒定结构域之间的柔性或半柔性连接。此外,本发明抗体的抗原结合片段可包含以上所列的任何可变结构域和恒定结构域构型的同源二聚体或异源二聚体(或其他多聚体),它们彼此非共价结合和/或与一个或多个单体vh或v
l
结构域(例如,通过二硫键)共价结合。与完整的抗体分子一样,抗原结合片段可以是单特异性或多特异性的(例如,双特异性的)。抗体的多特异性抗原结合片段通常包含至少两个不同的可变结构域,其中每个可变结构域均能够特异性结合单独的抗原或结合同一抗原上的不同表位。任何多特异性抗体形式,包括本发明公开的示例性双特异性抗体形式,均可使用本领域可用的常规技术,适于在本发明抗体的抗原结合片段上下文中使用。在本发明所述的某些实施方案中,本发明所述抗体是人抗体。
[0062]
一个或多个cdr残基的置换或一个或多个cdr的省略亦是可能的。抗体已在科学文献中进行描述,其中可省去一个或两个cdr来进行结合。padlan等人(1995faseb j.9:133-139)基于已发表的晶体结构分析了抗体与其抗原之间的接触区域,并得出结论,仅有大约1/5至1/3的cdr残基实际上与抗原接触。padlan等人还发现了许多抗体,其中一个或两个cdr不具有与抗原接触的氨基酸(亦参见vajdos et al.2002j mol biol 320:415-428)。
[0063]
不接触抗原的cdr残基可基于先前的研究(例如,通常无需cdrh2中的残基h60-h65),通过分子建模和/或凭经验从位于柯西亚(chothia)cdr之外的卡巴(kabat)cdr区域进行识别。如果省略cdr或其残基,通常用占据另一人抗体序列或此类序列的共有序列中相应位置的氨基酸进行置换。亦可凭经验选择cdr内的置换位置以及要置换的氨基酸。经验置换可以是保守置换或非保守置换。
[0064]
与相应的种系序列相比,本发明公开的全人抗流感-ha单克隆抗体可在所述重链和轻链可变结构域的框架和/或cdr区包含一个或多个氨基酸置换、插入和/或缺失。通过将本发明公开的氨基酸序列与可从例如公共抗体序列数据库获得的种系序列进行比较,可容易地确定此类突变。本发明包括抗体及其抗原结合片段,其衍生自本发明公开的任何氨基酸序列,其中一个或多个框架和/或cdr区内的一个或多个氨基酸突变为衍生抗体的种系序列的相应残基,或突变为另一个人种系序列的相应残基,或突变为相应种系残基的保守氨基酸置换(此类序列变化在本发明中统称为“种系突变”)。本领域普通技术人员从本发明公
开的重链和轻链可变区序列开始,可容易地生成许多抗体和抗原结合片段,所述抗体和抗原结合片段包含一个或多个个体种系突变或其组合。在某些实施方案,vh和/或v
l
结构域内的所有框架和/或cdr残基均突变回在衍生抗体的原始种系序列中发现的残基。在其他实施方案,仅有某些残基突变回原始种系序列,例如,仅在fr1的前8个氨基酸内或在fr4的最后8个氨基酸内发现的突变残基,或仅在cdr1、cdr2或cdr3中发现的突变残基。在其他实施方案,将一个或多个框架和/或cdr残基突变为不同种系序列(即,种系序列不同于最初衍生抗体的种系序列)的相应残基。此外,本发明所述抗体可包含框架和/或cdr区内的两个或更多个种系突变的任何组合,例如,其中某些个体残基突变为特定种系序列的相应残基,而某些与原始种系序列不同的其他残基保持或突变为不同种系序列的相应残基。一旦得到,可容易地测试含有一个或多个种系突变的抗体和抗原结合片段的一种或多种所需特性,例如改善的结合特异性,增加的结合亲和力,改善或增强的拮抗或激动生物学特性(视情况而定),降低的免疫原性等。以此种一般方式得到的抗体和抗原结合片段均包括在本发明中。
[0065]
本发明还包括全人抗流感-ha单克隆抗体,其包含具有一个或多个保守置换的本发明公开的hcvr、lcvr和/或cdr氨基酸序列中任一个的变体。譬如,本发明包括具有hcvr、lcvr和/或cdr氨基酸序列的抗流感-ha抗体,相对于本发明公开的任何hcvr、lcvr和/或cdr氨基酸序列,其具有例如10个或更少、8个或更少、6个或更少、4个或更少等的保守氨基酸置换。
[0066]
本发明使用的术语“人抗体”旨在包括具有衍生自人种系免疫球蛋白序列的可变区和恒定区的抗体。本发明的人mab可包括不由人种系免疫球蛋白序列编码的氨基酸残基(例如,通过体外随机或位点特异性诱变或通过体内体细胞突变而引入的突变),例如在cdr中,特别是在cdr3中。然而,本发明使用的术语“人抗体”不旨在包括其中衍生自另一种哺乳动物物种(例如小鼠)的种系的cdr序列已移植到人fr序列上的mab。所述术语包括在非人哺乳动物中或在非人哺乳动物的细胞中重组产生的抗体。所述术语不旨在包括分离自人受试者或在人受试者中产生的抗体。所述术语不包括在天然存在的、未经修饰的活生物体中通常不经修饰或人为干预/操作而存在的天然存在的抗体。
[0067]
本发明使用的术语“重组”是指抗体或其抗原结合片段,通过本领域已知的技术或方法作为重组dna技术而产生、表达、分离或获得,包括例如dna剪接和转基因表达。所述术语是指在非人哺乳动物(包括转基因非人哺乳动物(例如,转基因小鼠)或细胞(例如,cho细胞)表达系统中表达的抗体或从重组组合人抗体文库中分离的抗体。本发明使用的短语“重组人抗体”旨在包括通过重组方式制备、表达、产生或分离的所有人抗体,例如使用转染到宿主细胞中的重组表达载体表达的抗体;从重组、组合人抗体文库分离的抗体;从动物(例如小鼠)分离的抗体,所述动物是人免疫球蛋白基因的转基因动物(参见例如,taylor et al.(1992)nucl.acids res.20:6287-6295);或通过涉及将人免疫球蛋白基因序列剪接到其他dna序列的任何其他方法制备、表达、产生或分离的抗体。此类重组人抗体具有衍生自人种系免疫球蛋白序列的可变区和恒定区。然而,在某些实施方案,对此种重组人抗体进行体外诱变(或者,当使用转基因人ig序列的动物时,进行体内体细胞诱变),并因此重组抗体的所述vh区和v
l
区的氨基酸序列是衍生自人种系vh和v
l
序列并与之相关、但在体内可不天然存在于人抗体种系储库中的序列。人抗体可以与铰链异质性相关的两种形式存在。在一种形式中,免疫球蛋白分子包含约150-160kda的稳定四链构建体,其中二聚体通过链间重
链二硫键连接在一起。在第二种形式中,所述二聚体不通过链间二硫键连接,并且形成约75-80kda的分子,其由共价偶联的轻链和重链(半抗体)组成。即使在亲和纯化后,这些形式也极难分离。在各种完整igg同种型中出现第二种形式的频率是由于但不限于,与抗体的铰链区同种型相关的结构差异。人igg4铰链的铰链区中的单个氨基酸置换可显著地将第二种形式(angal et al.(1993)molecular immunology 30:105)的出现降低至通常使用人igg1铰链观察到的水平。本发明公开内容包括在铰链区,ch2区或ch3区中具有一个或多个突变的抗体,所述突变可能是例如在生成中所需的,以提高所需抗体形式的产量。
[0068]
本发明使用的“分离的抗体”旨在指基本上不含具有不同抗原特异性的其他抗体(ab)的抗体(例如,特异性结合流感-ha的分离的抗体或其片段,基本上不含特异性结合流感-ha以外的抗原的ab)。本发明所述抗体可以是分离的抗体。本发明使用的“分离的抗体”是指已经从其天然环境的至少一种组分中鉴定和分离和/或回收的抗体。例如,出于本发明目的,已从生物体的至少一种组分或从其中天然存在或天然产生的抗体的组织或细胞中分离或移去的抗体是“分离的抗体”。分离的抗体还包括重组细胞内的原位抗体。分离的抗体是已经过至少一个纯化或分离步骤的抗体。根据某些实施方案,分离的抗体可以基本上不含其他细胞材料和/或化学物质。与衍生抗体的相应种系序列相比,本发明使用的抗体可在所述重链和轻链可变结构域的框架和/或cdr区包含一个或多个氨基酸置换、插入和/或缺失。通过将本发明公开的氨基酸序列与可从例如公共抗体序列数据库获得的种系序列进行比较,可容易地确定此类突变。本发明包括抗体及其抗原结合片段,其衍生自本发明公开的任何氨基酸序列,其中一个或多个框架和/或cdr区内的一个或多个氨基酸突变为衍生抗体的种系序列的相应残基,或突变为另一个人种系序列的相应残基,或突变为相应种系残基的保守氨基酸置换(此类序列变化在本发明中统称为“种系突变”)。本领域普通技术人员从本发明公开的重链和轻链可变区序列开始,可容易地生成许多抗体和抗原结合片段,所述抗体和抗原结合片段包含一个或多个个体种系突变或其组合。
[0069]
本发明使用的“阻断抗体”、“中和抗体”或“拮抗剂抗体”旨在指一种抗体,其与抗原的结合使得抑制与所述抗原相关的至少一种生物活性。譬如,本发明抗体或抗体-药物偶联物可防止或阻断流感附着或进入宿主细胞。此外,“中和抗体”是可中和即预防、抑制、减少、阻碍或干扰病原体在宿主中引发感染和/或使感染持续的能力的抗体。此类抗体或抗体-药物偶联物,当通过与流感ha结合而具有中和能力时,可被称为“中和流感-ha活性的抗体”。术语“中和抗体”和“中和
……
的抗体”或“中和
……
的抗体类”在本发明中可互换使用。所述这些抗体可单独使用或与其他抗病毒剂在适当配制后作为预防剂或治疗剂联合使用,或与主动疫苗接种结合使用,或作为诊断工具。本发明使用的“抗流感抗体”可指一种抗体,其与抗原(例如ha)的结合使得抑制与流感病毒相关的至少一种生物活性。
[0070]
术语“表位”是指与抗体分子的可变区中被称为互补位的特定抗原结合位点相互作用的抗原决定簇。单个抗原可具有一个以上的表位。因此,不同的抗体可结合抗原上的不同区域并且可具有不同的生物学效应。术语“表位”还指抗原上b细胞和/或t细胞对其作出反应的位点。其还指被抗体结合的抗原区域。b细胞表位可由相邻接氨基酸形成或可由通过蛋白质的三级折叠并列的非相邻接氨基酸形成。由相邻接氨基酸形成的表位通常在暴露于变性溶剂时被保留,而由三级折叠形成的表位通常在用变性溶剂处理时丢失。表位通常包括至少3个,更通常是至少5个或8-10个具有独特空间构象的氨基酸。表位可定义为结构性
或功能性的。功能性表位通常是结构性表位的子集,并且具有直接有助于相互作用亲和力的那些残基。表位亦可以是构象性的,即由非线性氨基酸组成。在某些实施方案,表位可包括作为诸如氨基酸类、糖侧链类、磷酰基团类或磺酰基团类的分子的化学活性表面组的决定簇,并且在某些实施方案,可具有特定的三维结构特征,和/或特定的电荷特征。
[0071]
术语“表面等离子体共振”是指允许通过检测生物传感器基质内蛋白质浓度的变化,例如使用biacore
tm
系统,来分析实时生物分子相互作用的光学现象(pharmacia biosensor ab,uppsala,sweden和piscataway,n.j)。
[0072]
生物膜干涉测定法是用于测定生物分子相互作用的无标记技术。其是一种光学分析技术,可分析从两个表面反射的白光的干涉图谱:生物传感器尖端上的固定蛋白质层以及内部参考层。与生物传感器尖端结合的分子数量的任何变化均会导致可实时测定的干涉图谱的位移变化(abdiche,y.n.,et al.analytical biochemistry,(2008),377(2),209-217)。在某些实施方案,“基于实时生物膜干涉仪的生物传感器(octet htx测定)”用于评估某些抗流感ha抗体的结合特性。
[0073]
本发明使用的术语“k
d”旨在指特定抗体-抗原相互作用的平衡解离常数。
[0074]
本发明使用的短语“交叉竞争”是指一种抗体或其抗原结合片段结合抗原并抑制或阻断另一种抗体或其抗原结合片段的结合。所述短语还包括两种抗体之间在两个方向上的竞争,即第一抗体结合并阻断第二抗体的结合,反之亦然。在某些实施方案,所述第一抗体和第二抗体可结合相同的表位。或者,所述第一抗体和第二抗体可结合不同但重叠的表位,使得其中一个抗体的结合,例如通过空间位阻,抑制或阻断所述第二抗体的结合。抗体之间的交叉竞争可通过本领域已知的方法来测定,例如通过实时、无标记的生物膜干涉测定法来测定。两种抗体之间的交叉竞争可表现为由于自身结合而导致的所述第二抗体的结合小于背景信号(其中第一抗体和第二抗体均为相同的抗体)。譬如,两种抗体之间的交叉竞争可表示为小于基线自身背景结合的所述第二抗体的结合百分比(其中第一抗体和第二抗体均为相同的抗体)。
[0075]
当提及核酸或其片段时,术语“基本同一性”或“基本上相同”表示,当使适当的核苷酸插入或缺失与另一核酸(或其互补链)进行最佳比对时,通过任何众所周知的序列同一性算法(例如fasta、blast、或gap)测定,在至少约90%、更优选至少约95%、96%、97%、98%或99%的核苷酸碱基中存在核苷酸序列同一性,如wo 2016/100807或us 2016/0176953 a1中所讨论,其各自以全文引用的方式并入本发明。在某些实施例,与参比核酸分子具有基本同一性的核酸分子可编码具有与参比核酸分子编码的多肽相同或基本相似的氨基酸序列的多肽。
[0076]
当应用于多肽时,短语“基本相似性”或“基本上相似”是指两个肽序列在最佳比对时,例如通过程序gap或bestfit使用默认空位权重,共享至少90%的序列同一性,甚至更优选至少95%、98%或99%的序列同一性。优选地,不相同的残基位置因保守氨基酸置换而不同。“保守氨基酸置换”是其中氨基酸残基被具有相似化学性质(例如电荷或疏水性)的侧链(r基团)的另一氨基酸残基置换的氨基酸置换。通常,保守氨基酸置换不会显著改变蛋白质的功能特性。在通过保守置换使两个或更多个氨基酸序列彼此不同的情况下,可以向上调整序列同一性百分比或相似程度以校正所述置换的保守性质。进行此种调节的方法是本领域技术人员所熟知的。(参见例如,pearson(1994)methods mol.biol.24:307-331)。具有相
似化学性质的侧链的氨基酸基团的实例包括(1)脂族侧链:甘氨酸,丙氨酸,缬氨酸,亮氨酸和异亮氨酸;(2)脂肪族-羟基侧链:丝氨酸和苏氨酸;(3)含酰胺的侧链:天冬酰胺和谷氨酰胺;(4)芳香族侧链:苯丙氨酸,酪氨酸和色氨酸;(5)碱性侧链:赖氨酸,精氨酸和组氨酸;(6)酸性侧链:天冬氨酸和谷氨酸,和(7)含硫侧链:半胱氨酸和蛋氨酸。优选的保守氨基酸置换基团是:缬氨酸-亮氨酸-异亮氨酸,苯丙氨酸-酪氨酸,赖氨酸-精氨酸,丙氨酸-缬氨酸,谷氨酸-天冬氨酸和天冬酰胺-谷氨酰胺。或者,保守置换是在gonnet et al.(1992)science 256:1443-1445中公开的pam250对数似然矩阵(log-likelihood matrix)中具有正值的任何变化。“适度保守”置换是pam250对数似然矩阵中具有非负值的任何变化。
[0077]
通常使用序列分析软件来测定多肽的序列相似性。蛋白质分析软件使用分配给各种置换、缺失和其他修饰(包括保守氨基酸置换)的相似性测定来匹配相似的序列。例如,gcg软件包含诸如gap和bestfit程序,其可与默认参数一起使用以确定密切相关的多肽(例如来自不同生物物种的同源多肽)之间的序列同源性或序列同一性,或野生型蛋白质与其突变体之间的序列同源性或序列同一性。参见例如,gcg 6.1版本(gcg version 6.1)。也可采用fasta使用默认或推荐的参数(gcg version 6.1中的程序)来比较多肽序列。fasta(例如,fasta2和fasta3)提供查询和搜索序列之间最佳重叠区域的比对和百分比序列同一性(pearson(2000)同上)。序列亦可使用smith-waterman同源性搜索算法进行比较,所述算法使用空位开放罚分为12和空位延伸罚分为2以及blosum矩阵为62的仿射空位搜索。当将本发明序列与包含来自不同生物体的大量序列的数据库进行比较时,另一种优选算法是使用默认参数的计算机程序blast,尤其是blastp或tblastn。参见例如,altschul et al.(1990)j.mol.biol.215:403-410和altschul et al.(1997)nucleic acids res.25:3389-3402。
[0078]
短语“治疗有效量”是指产生经施用的所需效果的量。确切的量将取决于治疗目的,并且可由本领域技术人员使用已知技术确定(参见,例如,lloyd(1999)the art,science and technology of pharmaceutical compounding)。
[0079]
本发明使用的术语“受试者”是指需要改善、预防和/或治疗疾病或病症(例如病毒感染)的动物,优选哺乳动物,更优选人。受试者可能患有流感感染或倾向于发生流感病毒感染。“易患流感病毒感染”的受试者或“感染流感病毒的风险可能较高”的受试者是那些因自身免疫性疾病而免疫系统受损的受试者,那些接受免疫抑制治疗(例如,器官移植后)的人、那些患有人类免疫缺陷综合征(hiv)或获得性免疫缺陷综合征(aids)的人、消耗或破坏白细胞的某些类型的贫血症患者、那些接受放疗或化疗的人、或那些患有炎症性疾病的人。此外,极端年幼或年老的受试者面临的风险亦会增加。任何与受感染者有身体接触或身体接近的人,均会增加感染流感病毒的风险。此外,受试者由于接近疾病爆发而处于感染流感病毒的风险中,例如,受试者居住在人口稠密的城市或与已确证或疑似感染流感病毒的受试者非常接近,或工作选择,例如,医院工作人员、药物研究人员、前往感染地区的旅行者或飞行常客。
[0080]
本发明使用的术语“治疗”或“处理”是指由于向有需要的受试者施用治疗剂(例如,公开的抗体)而降低或改善流感感染的至少一种症状或适应症的严重程度。所述术语包括抑制疾病进展或感染恶化。所述术语还包括疾病的积极预后,即受试者在施用治疗剂(例如,公开的抗体或抗体-药物偶联物)后可能无感染或可能具有降低的病毒滴度或无病毒滴
度。治疗剂可以治疗剂量施用于受试者。
[0081]
术语“预防”、“防止”或“阻止”是指在施用所公开的抗体或抗体-药物偶联物后抑制流感感染的表现或抑制流感感染的任何症状或适应症。所述术语包括防止感染在暴露于病毒或处于感染流感风险的对象中传播。
[0082]
本发明使用的“保护作用”可通过本领域已知的任何标准程序来证明,以确定诸如抗病毒剂之类药剂、或诸如抗流感-ha抗体之类的抗体、或本发明公开的抗体-药物偶联物是否可证明以下任何一种或多种:譬如,暴露于传染原后存活率增加、病毒载量降低、或与传染原相关的至少一种症状得到改善。
[0083]
本发明使用的短语“抗病毒药物”、“抗病毒”、“抗病毒化合物”和“抗病毒类化合物”适用于用于治疗、预防或改善受试者的病毒感染(例如,流感感染)的抗感染药物或疗法。术语“抗病毒药物”(或其同义词“抗病毒类药物”、“抗病毒化合物”和“抗病毒类化合物”)包括但不限于,(奥司他韦)、(扎那米韦)、利巴韦林、或干扰素-α2b。抗病毒类药物包括流感抑制剂类。本发明使用的“流感抑制剂”是指用于抑制流感病毒感染的药物,包括但不限于奥司他韦。本发明使用的聚合酶抑制剂可指核酸聚合酶(例如,流感聚合酶)的抑制剂。示例性聚合酶抑制剂是vx-787。不希望受任何特定理论的束缚,流感抑制剂可通过靶向流感病毒本身或通过靶向可能被流感病毒靶向的宿主细胞来发挥作用。譬如,靶向宿主细胞的流感抑制剂可抑制细胞中的翻译,从而减少病毒复制。
[0084]
短语“特异性结合”或“与
……
特异性结合”或诸如此类,是指抗体或其抗原结合片段与在生理条件下相对稳定的抗原形成复合物。特异性结合可通过至少约1x10-8 m或更小的平衡解离常数来表征(例如,较小的kd表示结合更紧密)。用于确定两个分子是否特异性结合的方法是本领域众所周知的并且包括例如平衡透析法、表面等离子体共振等。如本发明所述,抗体已通过在htx生物传感器上的实时、无标记生物膜干涉测定法进行识别,其特异性结合流感-ha。此外,与流感-ha中的一个结构域以及一种或多种额外抗原结合的多特异性抗体或与流感-ha的两个不同区域结合的双特异性抗体仍然被认为是“特异性结合”的抗体(如本发明所用)。除了中和抗体,特异性结合ha但非中和的抗体亦可涵盖在本发明范围内用于生成抗体-药物偶联物。此类抗体可发挥作用,例如,将有效负载递送至受感染流感的细胞。
[0085]
术语“高亲和性”或“高亲和力”抗体是指对流感-ha具有至少10-8
m,优选10-9
m,更优选10-10
m,甚至更优选10-11
m,甚至更优选10-12
m的结合亲和性的那些mab,所述结合亲和性以kd表示,通过实时、无标记生物膜干涉测定法(例如,htx生物传感器)、或通过表面等离子体共振(例如,biacore
tm
)、或通过溶液亲和性elisa进行测定。
[0086]
短语或术语“解离速率”、“k
off”或“kd”是指从流感-ha解离的抗体,其速率常数为1x10-3 s-1
或更小,优选1x10-4 s-1
或更小,通过实时、无标记生物膜干涉测定法(例如,htx生物传感器)、或通过表面等离子体共振(例如,biacore
tm
)进行测定。
[0087]
本发明使用的短语抗体的“抗原结合结构域”或“抗原结合部分”、抗体的“抗原结合片段”,诸如此类,包括任何天然存在的、酶促获得的、合成的、或基因工程化的多肽或糖蛋白,其特异性结合抗原以形成复合物。
[0088]
在具体实施方案中,本发明的抗体或抗体片段可偶联至基团部分,例如配体或治疗性基团部分(“抗体-药物偶联物”或“免疫偶联物”),例如抗病毒药物、包括抗病毒药物的
连接体-有效负载、第二抗流感抗体、或可用于治疗由流感-ha引起的感染的任何其他治疗基团部分。
[0089]
本发明使用的“依次施用”是指在不同的时间点,例如在以预定间隔(例如,小时、天、周、或月)隔开的不同天数,将化合物的每个剂量施用于受试者。
[0090]
短语“初始剂量”,“二次剂量”,和“三次剂量”是指本发明所述化合物的给药时间顺序。因此,“初始剂量”是在治疗方案开始时给药的剂量(也称为“基线剂量”);“二次剂量”是在所述初始剂量给药后施用的剂量;和“三次剂量”是在所述二次剂量给药后施用的剂量。所述初始、二次和三次剂量均可含有相同量的本发明所述化合物,但通常可在给药频率方面彼此不同。
[0091]
本发明使用的短语“紧随前述剂量”是指在多次给药的序列中,在施用紧邻下一个剂量之前,对患者施用本发明化合物的剂量,在所述给药序列中不存在干预剂量。
[0092]
本发明使用的术语“有效负载”是指小分子活性成分(例如,抗病毒化合物),任选地直接或经由连接体偶联至抗体或其抗原结合片段,以提供所需生物学效应(例如,抑制流感病毒感染或复制)。有效负载可小于或等于2,000da,小于或等于1,500da,或小于或等于900da。化合物或有效负载
[0093]
本发明提供了抗病毒化合物或有效负载。不受任何特定操作理论的束缚,所述抗病毒化合物包括(1)vx-787及其衍生物;(2)巴洛沙韦(baloxavir)及其衍生物(例如,巴洛沙韦酯(baloxavir marboxil))。在某些实施方案,所述抗病毒化合物可作为偶联物的一部分递送至细胞。在某些实施方案,所述抗病毒化合物能够在靶标(例如,靶细胞)处或靶标(例如,靶细胞)中发挥vx-787、巴洛沙韦和/或巴洛沙韦酯及其每一种衍生物的任何活性。某些抗病毒化合物可具有一种或多种附加活性。
[0094]
在某些实施方案,本发明提供了具有式i所示结构的化合物:或其药学上可接受的盐。在式i中,在某些实施方案,zz是
–
or1或
–
nhoh。在一个实施方案,zz是
–
nhoh。在一个实施方案,zz是
–
or1。在式i中,当zz是
–
or1时,有用的r1基团包括h和在一个实施方案,r1是h。在一个实施方案,r1是在一个实施方
案,所述化合物是vx-787或
[0095]
在某些实施方案,本发明提供了具有式ii所示结构的化合物:或其药学上可接受的盐。在一个实施方案,r1是h。在一个实施方案,r1是
–
ch2oc(o)och3。在一个实施方案,r1是在一个实施方案,所述化合物是巴洛沙韦酯(baloxavir marboxil)或在一个实施方案,所述化合物是巴洛沙韦或结合剂
[0096]
可用于本发明提供的任何偶联物的适宜结合剂包括但不限于,抗体、病毒受体、或任何其他细胞结合分子类或物质类或肽结合分子类或物质类。示例性流感ha的全长氨基酸序列示出在genbank中,登录号为acp44150.1。
[0097]
合适的结合剂包括特异性结合流感病毒蛋白(例如,表面蛋白血凝素(ha)、神经氨酸酶(na)和matrix-2(m2))的抗体(例如,全人抗体)及其抗原结合片段。在一些实施方案,所述这些结合剂调节流感病毒与宿主细胞的相互作用。在一些实施方案,所述抗体或其抗
原结合片段结合成熟血凝素。在一些实施方案,所述抗体或其抗原结合片段结合ha0血凝素前体蛋白。所述抗流感ha抗体可以高亲和性结合流感病毒ha。在某些实施方案,本发明抗体是阻断抗体,其中所述抗体可结合流感ha并阻断病毒附着和/或进入宿主细胞。在一些实施方案,本发明的阻断抗体可阻断流感病毒与细胞的结合,因此可抑制或中和宿主细胞的病毒感染性。在一些实施方案,所述阻断抗体可用于治疗患有流感病毒感染的受试者。当将所述抗体施用于有需要的受试者时,可减少所述受试者被流感病毒等病毒感染。其可用于降低受试者的病毒载量。其可单独使用或与本领域已知的用于治疗病毒感染的其他治疗性基团部分或方式一同用作辅助疗法。在某些实施方案,所述这些抗体可与病毒ha的茎区中的表位、病毒ha的头部区中的表位或此两者中的表位结合。此外,所识别的抗体可进行预防性使用(感染前)以保护哺乳动物免受感染,或可进行治疗性使用(感染建立后)以改善先前建立的感染,或改善与所述感染相关的至少一种症状。
[0098]
在某些实施方案,所述抗体获自用初级免疫原例如用全长流感ha或用流感ha或其片段的重组形式免疫,然后用次级免疫原免疫或用流感ha的免疫原性活性片段免疫的小鼠。在某些实施方案,所述抗体获自用流感疫苗组合物免疫,然后用一种或多种重组生成的ha肽加强免疫的小鼠。在某些实施方案,所述抗体获自人类。在某些实施方案,所述抗体获自哺乳动物类(例如,非人哺乳动物类)。在某些实施方案,所述抗体获自非人灵长类动物。
[0099]
免疫原可以是流感ha的生物活性和/或免疫原性片段或编码其活性片段的dna。所述片段可来源于ha蛋白的茎区(参见,sui et al.nature struct,and mol.biol.published online 22feb.2009;pages 1-9)、ha蛋白的头部区域、或其组合。
[0100]
可对肽进行修饰以包括添加或置换某些残基以用于标记或用于与载体分子(例如,钥孔血蓝蛋白(klh))偶联的目的。譬如,可在肽的n端或c端添加半胱氨酸,或者可添加连接体序列以制备所述肽,其用于偶联至例如用于免疫的klh。
[0101]
如通过体外或体内测定法所测定,本发明的某些抗流感抗体、抗流感-ha抗体或adc均具有抗病毒活性,例如能够结合以及中和流感-ha的活性。如体外或体内测定法所测定,本发明的某些抗流感抗体、抗流感-ha抗体或adc均能够结合ha但不具有中和活性。本发明的抗体或adc结合以及中和流感-ha的活性并因此所述病毒附着和/或进入宿主细胞并随后发生病毒感染的能力,可使用本领域技术人员已知的任何标准方法进行测定,包括如本发明所述的结合测定法或活性测定法。
[0102]
流感-ha特异性抗体或adc可不包含附加标记或基团部分,或者其也可包含n端或c端标记或基团部分。在一个实施方案,所述标记或基团部分是生物素。在结合测定试验中,标记(若有)的位置可确定肽相对于肽结合的表面的方向。譬如,如果表面涂有抗生物素蛋白,则含有n端生物素的肽将被定向,使得所述肽的c端部分处于所述表面的远端。在一个实施方案,所述标记可以是放射性核素、荧光染料、或mri可检测标记。在某些实施方案,此类经标记的抗体可用于诊断测定法,包括成像测定法。在一个实施方案,所述附加基团部分是肽标签。在一个实施方案,adc包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签。在一个实施方案,adc包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签(例如,五肽),其中所述肽标签是五肽序列llqga(例如,用于通过转谷氨酰胺酶偶联连接体-有效负载)。在一个实施方案,adc包括两条抗体重链并且进一步包括位于每条抗体重链c端的肽标签。在一个实施方案,adc包括两条抗体重链并且进一步包括位于每条
抗体重链c端的肽标签,其中所述肽标签是五肽序列llqga。
[0103]
在某些实施方案,所述抗体包含轻链。在某些实施方案,所述轻链是κ轻链。在某些实施方案,所述轻链是λ轻链。在某些实施方案,所述抗体包含重链。在一些实施方案,所述重链是iga。在一些实施方案,所述重链是igd。在一些实施方案,所述重链是ige。在一些实施方案,所述重链是igg。在一些实施方案,所述重链是igm。在一些实施方案,所述重链是igg1。在一些实施方案,所述重链是igg2。在一些实施方案,所述重链是igg3。在一些实施方案,所述重链是igg4。在一些实施方案,所述重链是iga1。在一些实施方案,所述重链是iga2。
[0104]
在一些实施方案,所述抗体是抗体片段。在一些实施方案,所述抗体片段是fv片段。在一些实施方案,所述抗体片段是fab片段。在一些实施方案,所述抗体片段是f(ab
′
)2片段。在一些实施方案,所述抗体片段是fab
′
片段。在一些实施方案,所述抗体片段是scfv(sfv)片段。在一些实施方案,所述抗体片段是scfv-fc片段。
[0105]
在一些实施方案,所述抗体是单克隆抗体。在一些实施方案,所述抗体是多克隆抗体。在一些实施方案,所述抗体是双特异性抗体,包括第一抗原结合结构域和第二抗原结合结构域。
[0106]
在一些实施方案,所述抗体是嵌合抗体。在一些实施方案,所述抗体是人源化抗体。在一些实施方案,所述抗体是人抗体。
[0107]
在某些实施方案,所述抗体在eu编号系统中编号为295的一个或多个重链位置处包含谷氨酰胺残基。在本发明中,此位置称为谷氨酰胺295,或称为gln295,或称为q295。技术人员将认识到,其是许多抗体的野生型序列中的保守谷氨酰胺残基。残基q295的鉴定可采用标准的序列比对工具容易地完成,包括本发明所述的那些。在其他有用的实施方案中,可将抗体设计成包含谷氨酰胺残基。在某些实施方案,所述抗体包含一个或多个n297q突变。修饰抗体序列以包括谷氨酰胺残基的技术均属于本领域技术人员的技术范围内(参见,例如ausubel et al.current protoc.mol.biol.)。在一个实施方案,所述抗体包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签。在一个实施方案,所述抗体包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签,例如,转谷氨酰胺酶识别序列或五肽标签,其中所述肽标签是五肽序列llqga。人抗体的制备
[0108]
在转基因小鼠中生成人抗体的方法均是本领域已知的。任何此类已知方法均可在本发明上下文中用于制备特异性结合流感-ha的人抗体。包含以下任一种的免疫原可用于生成针对流感ha的抗体。在某些实施方案,本发明抗体获自用全长天然流感ha(参见,例如,genbank登录号fj966082.1)、或用减毒或灭活的活病毒、或用编码蛋白质或其片段的dna进行免疫的小鼠。或者,流感-ha蛋白或其片段可使用标准生化技术生成并进行修饰并用作免疫原。在一个实施方案,所述免疫原是重组生成的流感-ha蛋白或其片段。在本发明的某些实施方案中,免疫原可以是流感病毒疫苗。在某些实施方案,可施用一次或多次加强注射。在某些实施方案,所述加强注射可包含一种或多种流感病毒株,或源自所述这些毒株的血凝素,例如,参见protein sciences的h1 a/new caledonia/20/1999、h5 a/lndonesia/05/2005、h3 a/victoria/361/2011、h7 a/netherlands/219/2003、或h9 a/hong kong/1073/1988、或乙型流感病毒株b/victoria/2/87、b/nanchang/3451/93、b/singapore/11/1994、
b/florida/4/2006、或b/yamagata/16/88。在某些实施方案,所述加强注射可包含流感毒株的1:1混合物,或源自所述毒株的血凝素的1:1混合物。在某些实施方案,所述免疫原可以是在大肠杆菌或任何其他真核或哺乳动物细胞如中国仓鼠卵巢(cho)细胞或流感病毒本身中表达的重组流感ha肽。
[0109]
使用技术(参见,例如us 6,596,541,regeneron pharmaceuticals,)或用于生成单克隆抗体的任何其他已知方法,最初分离的针对流感-ha的高亲和性嵌合抗体具有人可变区以及小鼠恒定区。技术涉及生成具有基因组的转基因小鼠,所述基因组包含与内源性小鼠恒定区基因座可操作连接的人重链和轻链可变区,使得小鼠产生包含人可变区以及小鼠恒定区以对抗原刺激作出反应的抗体。将编码抗体重链和轻链可变区的dna分离并与编码人重链和轻链恒定区的dna可操作地连接。然后在能够表达全人抗体的细胞中表达所述dna。
[0110]
通常,用目的抗原攻击小鼠,并从表达抗体的小鼠回收淋巴细胞(例如,b细胞)。可将淋巴细胞与骨髓瘤细胞系融合以制备永生杂交瘤细胞系,并且筛选和选择此类杂交瘤细胞系以识别可生成对目的抗原特异的抗体的杂交瘤细胞系。可分离编码重链和轻链可变区的dna并将其连接至所述重链和轻链的所需同种型恒定区。此种抗体蛋白可在细胞(例如,cho细胞)中生成。或者,编码所述抗原特异性嵌合抗体或所述轻链和重链可变结构域的dna可直接从抗原特异性淋巴细胞分离得到。
[0111]
最初,分离具有人可变区以及小鼠恒定区的高亲和性嵌合抗体。如在wo 2016/100807或us2016/0176953a1中所述,其各自均以全文引用方式并入,对所述抗体进行表征和选择以获得所需特征,包括亲和性、选择性、表位等。将小鼠恒定区替换为所需的人恒定区以生成本发明的全人抗体,例如,野生型或经修饰的igg1或igg4。虽然所选择的恒定区可根据具体用途而变化,但高亲和性抗原结合及靶标特异性特征均存在于可变区中。生物等效物
[0112]
本发明的抗流感-ha抗体和抗体片段均包括具有氨基酸序列的蛋白质,所述氨基酸序列不同于所述抗体的氨基酸序列但保留结合流感ha的能力。当与亲本序列相比时,此类变体抗体和抗体片段均包含氨基酸的一个或多个添加、缺失或置换,但却表现出基本上等同于所述抗体的生物学活性的生物学活性。同样,与所公开的序列相比,本发明公开的编码抗体的dna序列包括此类序列,所述序列包含核苷酸的一个或多个添加、缺失或置换,但编码基本上与本发明抗体或抗体片段生物等效的抗体或抗体片段。其他生物等效的抗流感-ha抗体和抗体片段均如wo 2016/100807或us 2016/0176953 a1中所述,其各自均以全文引用方式并入。抗体的生物学特性
[0113]
通常,本发明抗体通过与流感ha结合而发挥作用。譬如,本发明提供了结合流感ha(例如,在25℃或37℃)的抗体及抗体的抗原结合片段,通过基于实时生物膜干涉仪的生物传感器(octet htx测定)或通过表面等离子体共振进行测定,其kd小于10nm。在某些实施方案,通过表面等离子体共振进行测定,例如,使用如wo 2016/100807或us 2016/0176953 a1中所述的测定形式,各专利文献均以全文引用方式并入,或使用基本上相似的测定法,所述抗体或其抗原结合片段以小于约5nm、小于约2nm、小于约1nm、小于约500pm、小于250pm、或小于100pm的kd结合流感-ha。
[0114]
用于测定结合活性的非限制性示例性体外测定法示出在wo 2016/100807或us 2016/0176953a1的实施例3中,各专利文献均通过引用其全文并入本发明。在wo 2016/100807或us 2016/0176953 a1的实施例3中,抗流感-ha抗体对流感-ha的结合亲和性和解离常数均通过基于实时生物膜干涉仪的生物传感器(octet htx测定)进行测定。在wo 2016/100807或us 2016/0176953 a1的实施例4和实施例5中,中和测定法用于测定不同的组1流感病毒株的感染性。在wo 2016/100807或us 2016/0176953 a1的实施例6中,某些抗体显示出在体外介导病毒感染细胞的补体依赖性细胞毒性(cdc)。wo 2016/100807或us 2016/0176953 a1的实施例7和实施例10表明,当本发明的某些抗体进行预防性或治疗性施用时,其均能够在体内中和甲型流感感染。
[0115]
本发明亦提供了结合流感ha的抗体及其抗原结合片段,通过25℃下的表面等离子体共振进行测定,例如,使用如wo 2016/100807或us 2016/0176953 a1中定义的测定形式,各专利文献均以全文引用方式并入本发明,或使用基本上相似的测定法,所述抗体及其抗原结合片段结合流感ha的解离半衰期(t1/2)大于约100分钟。在某些实施方案,通过25℃下的表面等离子体共振进行测定,例如,使用如wo 2016/100807或us 2016/0176953 a1中定义的测定形式,各专利文献均以全文引用方式并入本发明(例如,imab-捕获或抗原-捕获形式),或使用基本上相似的测定法,本发明抗体或抗原结合片段结合流感ha的t1/2大于约200分钟、大于约300分钟、大于约400分钟、大于约500分钟、大于约600分钟、大于约700分钟、大于约800分钟、大于约900分钟、或大于约1000分钟。在一个实施方案,本发明抗体和抗原结合片段以大于300分钟的解离半衰期(t1/2)结合流感ha。在一个实施方案,当在猴和小鼠中进行测试时,与指定为对照i mab的比较例抗体相比,本发明抗体提供了约1.5至2倍的解离半衰期增加。
[0116]
本发明还提供了可中和流感病毒对其宿主细胞的感染性的抗体或其抗原结合片段。在一些实施方案,在微量中和测定试验中,例如,如wo 2016/100807或us 2016/0176953 a1的实施例4和实施例5所示,各专利文献均以全文引用方式并入本发明,或使用基本上相似的测定法,所述抗体表现出针对各种代表性组1流感病毒(h1n1 a/puerto rico/08/1934;h5n1 a/vietnam/1203/2004;h1n1 a california/07/2009;h1n1 a/wisconsin/1933;h1n1 a/brisbane/59/1997,h9n2 a hong kong/33982/2009,h13n6 a/gull/maryland/704/1977和h16n3 a/shorebird/delaware/172/2006)的中和效力,其ic
50
范围为约1.6nm至约130nm。在一个实施方案,可中和流感病毒对其宿主细胞的感染性的抗体或其抗原结合片段的ic
50
小于130nm。
[0117]
本发明还提供了可介导受感染细胞的补体依赖性细胞毒性的抗体或其抗原结合片段,其ec
50
范围为约20nm至约66nm(参见wo 2016/100807或us 2016/0176953 a1中的实施例6,各专利文献均以全文引用方式并入发明)。在一个实施方案,所述抗体或其抗原结合片段介导受感染细胞的补体依赖性细胞毒性,其ec
50
小于66nm。
[0118]
本发明描述了抗甲型流感ha抗体,与对照抗体相比,其表现出在体内对甲型流感感染的保护或中和增强。某些抗体在预防性(感染前)或治疗性(感染后)给药时显示出中和作用;参见wo 2016/100807或us 2016/0176953 a1中的实施例7,各专利文献均以全文引用方式并入发明。
[0119]
在一个实施方案,本发明提供了分离的重组抗体或其抗原结合片段,其与流感ha
特异性结合,其中所述抗体或其片段表现出两种或更多种以下特性:(a)是全人单克隆抗体;(b)在表面等离子体共振测定法中测定,以小于10-9
m的解离常数(kd)结合流感ha;(c)表现出从约370分钟至大于1000分钟的解离半衰期(t1/2);(d)表现出对选自h1n1、h5n1、h9n2、h13n6和h16n3的组1甲型流感病毒具有中和作用,其ic
50
范围为约1.6nm至约130nm;(e)表现出补体介导的流感病毒感染细胞裂解,其ec
50
为约20nm至约66nm;或(f)表现出保护作用,通过在病毒攻击之前或之后给药时流感病毒感染动物模型中的存活率增加来测定。
[0120]
本发明抗体可具有两种或更多种上述生物学特性,或其任何组合。通过回顾包括本发明工作实施例的本
技术实现要素:
,本发明抗体的其他生物学特性对于本领域普通技术人员而言将均是显而易见的。重链和轻链可变区氨基酸及核苷酸序列
[0121]
在一些实施方案,与连接体-有效负载或有效负载偶联的抗体或其抗原结合片段可以是靶向流感ha的抗体。示例性流感ha抗体可在例如wo 2016/100807或us 2016/0176953 a1中获悉,各专利文献均以全文引用方式并入本发明。在一些实施方案,流感ha抗体包含:重链互补决定区(hcdr)-1,其包含seq id no:20;hcdr2,其包含seq id no:22;hcdr3,其包含seq id no:24;轻链互补决定区(lcdr)-1,其包含seq id no:28;lcdr2,其包含seq id no:30;和lcdr3,其包含seq id no:32。在一些实施方案,流感ha抗体包含:包含seq id no:18的重链可变区(hcvr)和包含seq id no:26的轻链可变区(lcvr)。在任一前述实施方案,所述流感ha抗体可通过定点诱变以在位点插入谷氨酰胺残基而不导致抗体功能或结合失效来制备得到。譬如,在任一前述实施方案,所述流感ha抗体可包含asn297gln(n297q)突变。此类具有n297q突变的抗体还可在其可变区包含一个或多个额外的天然存在的谷氨酰胺残基,转谷氨酰胺酶可接近所述这些残基并因此能够偶联至有效负载或连接体-有效负载。在一个实施方案,所述抗体包括hcvr并且进一步包括位于所述hcvr的所述c端的肽标签。在一个实施方案,所述抗体包括hcvr并且进一步包括位于所述hcvr的所述c端的肽标签,其中所述肽标签是五肽序列llqga。在一个实施方案,所述抗体包括两个hcvr并且进一步包括位于每个hcvr的所述c端的肽标签。在一个实施方案,所述抗体包括两个hcvr并且进一步包括位于所述hcvr的所述c端的肽标签,其中所述肽标签是五肽序列llqga。
[0122]
表1列出了所选抗流感ha抗体的重链和轻链可变区以及cdr的氨基酸序列标识符。表2列出了相应的核酸序列标识符。表1:氨基酸序列标识符
*mab在所述恒定区含有一个或多个突变。表2:核酸序列标识符
*mab在所述恒定区含有一个或多个突变。
[0123]
结合剂连接体可通过抗体或抗原结合分子内特定氨基酸处的附接而与结合剂(例如,抗体或抗原结合分子)连接。可在本发明此实施方案的上下文中使用的示例性氨基酸附接包括,例如,赖氨酸(参见例如,us 5,208,020;us 2010/0129314;hollander et al.,bioconjugate chem.,2008,19:358
–
361;wo 2005/089808;us 5,714,586;us 2013/0101546;和us 2012/0585592),半胱氨酸(参见例如,us 2007/0258987;wo 2013/055993;wo 2013/055990;wo 2013/053873;wo 2013/053872;wo 2011/130598;us 2013/0101546;和us 7,750,116),硒代半胱氨酸(参见例如,wo 2008/122039;和hofer et al.,proc.natl.acad.sci.,usa,2008,105:12451
–
12456),甲酰甘氨酸(参见例如,carrico et al.,nat.chem.biol.,2007,3:321
–
322;agarwal et al.,proc.natl.acad.sci.,usa,2013,110:46
–
51,和rabuka et al.,nat.protocols,2012,10:1052
–
1067),非天然氨基酸(参见例如,wo 2013/068874,和wo 2012/166559),和酸性氨基酸(参见例如,wo 2012/05982)。连接体也可通过与碳水化合物的附接而与抗原结合蛋白偶联(参见例如,us2008/0305497,wo 2014/065661,ryan et al.,food&agriculture immunol.,2001,13:127
–
130,和jeger et al.,angew chem int ed engl.,2010,49:9995-9997)。
[0124]
在一些实施例,所述结合剂是抗体或抗原结合分子,和所述抗体通过赖氨酸残基与所述连接体连接。在一些实施方案,所述抗体或抗原结合分子通过半胱氨酸残基与所述连接体连接。
[0125]
连接体可通过基于转谷氨酰胺酶的化学-酶促偶联法偶联至一个或多个谷氨酰胺残基(参见例如,jeger et al.,angew chem int ed engl.,2010,49:9995-9997和dennler et al.,bioconjugate chem.2014,25:569-578)。例如,在转谷氨酰胺酶存在下,可将抗体的一个或多个谷氨酰胺残基偶联至伯胺化合物。伯胺化合物包括,例如,有效负载或连接体-有效负载,其通过转谷氨酰胺酶介导的偶联直接提供抗体-药物偶联物。伯胺化合物还包括被活性基团官能化的连接体和间隔基团,这些基团随后可与其他化合物反应以合成抗体-药物偶联物。包含谷氨酰胺残基的抗体可从天然来源分离得到或经工程改造以包含一个或多个谷氨酰胺残基。用于将谷氨酰胺残基改造成抗体多肽链(经谷氨酰胺酰基修饰的抗体或抗原结合分子)的技术均包含在本领域从业者的技术范围内。在某些实施方案,抗体是无糖基化的。
[0126]
在某些实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子在至少一个多肽链序列中包含至少一个谷氨酰胺残基。在某些实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子包含两个重链多肽,其中每个具有一个gln295或q295残基。在进一步的实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子在重链295以外的位点处包含一个或多个谷氨酰胺残基。本发明包括带有本发明所述n297q突变的本章节抗体。
[0127]
在一个实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签。在一个实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签,其中所述肽标签是五肽序列llqga。在一个实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子包括两条抗体重链并且进一步包括位于每条抗体重链的所述c端的肽标签。在一个实施方案,所述抗体或经谷氨酰胺酰基修饰的抗体或抗原结合分子包括两条抗体重链并且进一步包括位于每条抗体重链的所述c端的肽标签,其中所述肽标签是五肽序列llqga。连接体
[0128]
在某些实施方案,本发明所述偶联物的连接体l部分是一个基团部分,例如二价基团部分,其将结合剂共价连接至本发明所述的有效负载化合物。在其他实施例,连接体l是将结合剂共价连接至本发明所述的有效负载化合物的三价或多价基团部分。合适的连接体可在例如antibody-drug conjugates and immunotoxins;phillips,g.l.,ed.;springer verlag:new york,2013;antibody-drug conjugates;ducry,l.,ed.;humana press,2013;antibody-drug conjugates;wang,j.,shen,w.-c.,and zaro,j.l.,eds.;springer international publishing,2015中获悉,各文献的内容均通过引用其全文并入本发明。在某些实施方案,本发明所述连接体-有效负载的连接体l部分是与本发明所述的有效负载化合物共价连接的基团部分,其能够将结合剂二价连接及共价连接至本发明所述的有效负载化合物。在其他实施例,本发明所述的连接体-有效负载的连接体l部分是与本发明所述的有效负载化合物共价连接的基团部分,其能够作为三价或多价基团部分,将结合剂共价连
接至本发明所述的有效负载化合物。有效负载化合物包括上述式i和ii所示化合物,以及其与连接体l连接或掺入后的残基是连接体-有效负载。所述连接体-有效负载可进一步与结合剂例如抗体或其抗原结合片段连接以形成抗体-药物偶联物。本领域技术人员将认识到,有效负载基团部分的某些官能团便于连接至连接体和/或结合剂。例如,在某些实施方案,不存在连接体,并且有效负载直接连接至结合剂。在另一实施方案,有效负载包括羧酸,并且结合剂包括赖氨酸,其中各羧酸和赖氨酸均参与酰胺键形成以将有效负载残基直接连接至结合剂残基。有效负载官能团进一步包括羟基(例如,巴洛沙韦(baloxavir),及其衍生物,例如巴洛沙韦酯(baloxavir marboxil)衍生物)和羧酸(例如,在与l连接时呈酯形式,如在vx-787及其衍生物中所示)。
[0129]
在某些实施方案,所述连接体在生理条件下是稳定的。在某些实施方案,所述连接体是可切割的,例如,能够在酶存在下或在特定ph范围或ph值下释放至少所述有效负载部分。在一些实施方案,连接体包含酶可切割的基团部分。示例性酶可切割的基团部分包括但不限于肽键类、酯键类、腙类、和二硫键类。在一些实施方案,所述连接体包含组织蛋白酶可切割的连接体。
[0130]
在一些实施方案,所述连接体包含不可切割基团部分。在一些实施方案,所述不可切割的连接体衍生自或其残基。在一些实施方案,所述不可切割的连接体-有效负载残基是或其区域异构体。在一些实施方案,所述不可切割的连接体衍生自或其残基。在一些实施方案,所述不可切割的连接体-有效负载残基是或其区域异构体。在一个实施方案,所述连接体是马来酰亚胺环己烷羧酸酯或4-(n-马来酰亚胺基甲基)环己烷羧酸(mcc)。在所述结构中,表示与结合剂连接的键。在所述结构中,在一些实施例,表示由例如具有叠氮化物或炔烃官能团的结合剂与具有互补炔烃或叠氮化物官能团的连接体-有效负载的反应而产生的点击化学残基。在所述结构中,在其他实施例,表示二价硫化物,其由例如一种或多种结合剂半胱氨酸与一种或多种具有马来酰亚胺官能团的连接体或连接体-有效负载经由迈克尔加成反应的反应而产生。在所述结构中,在其他实施例,表示由例如一种或多种结合剂赖氨酸与一种或多种具有活化或未经活化羧基官能团的连接体或连接体-有效负载的反应而产生的酰胺键,如本领域技术人员所理解的。在
一个实施方案,表示由例如一种或多种结合剂赖氨酸与一种或多种具有活化羧基官能团的连接体或连接体-有效负载的反应而产生的酰胺键,如本领域技术人员所理解的。
[0131]
在一些实施方案,合适的连接体包括但不限于与单一结合剂(例如抗体)的两个半胱氨酸残基进行化学键合的那些。此类连接体可用于模拟由于偶联过程而被破坏的所述抗体的二硫键。
[0132]
在一些实施方案,所述连接体包含一个或多个氨基酸。合适的氨基酸包括天然、非天然的、标准、非标准的、蛋白原、非蛋白原的、和l-型或d-型α-氨基酸。在一些实施方案,所述连接体包含丙氨酸、缬氨酸、甘氨酸、亮氨酸、异亮氨酸、蛋氨酸、色氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、半胱氨酸、酪氨酸、天冬酰胺、谷氨酰胺、天冬氨酸、谷氨酸、赖氨酸、精氨酸、组氨酸、或瓜氨酸、或其衍生物、或其任何组合(例如,二肽类、三肽类、寡肽类、多肽类等)。在某些实施方案,所述氨基酸的一个或多个侧链连接至如下所述的侧链基团。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:缬氨酸和瓜氨酸(例如,二价-val-cit-或二价-vcit-)。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:丙氨酸和丙氨酸,或二价
–
aa
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:谷氨酸和丙氨酸,或
–
ea
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:谷氨酸和甘氨酸,或
–
eg
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:甘氨酸和甘氨酸,或
–
gg
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:谷氨酰胺、缬氨酸和瓜氨酸,或
–
q-v-cit
–
或
–
qvcit
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:谷氨酸、缬氨酸和瓜氨酸,或
–
e-v-cit
–
或
–
evcit
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:
–
ggggs
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:
–
ggggg
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:
–
ggggk
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:
–
gfgg
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:赖氨酸、缬氨酸和瓜氨酸,或
–
kvcit
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:
–
kva
–
。在一些实施方案,所述连接体是包含下列氨基酸或由下列氨基酸组成的肽:
–
va
–
。如本领域技术人员将理解的,在本段的任一实施方案中,并贯穿本发明,使用标准的三字母或单字母氨基酸命名。示例性单字母氨基酸名称包括,g代表甘氨酸,k代表赖氨酸,s代表丝氨酸,v代表缬氨酸,a代表丙氨酸,以及f代表苯丙氨酸。
[0133]
在一些实施方案,所述连接体包含自降解(self-immolative)基团。所述自降解基团可以是技术人员已知的任何此类基团。在特定实施方案,所述自降解基团是对氨基苄基(pab)或其衍生物。有用的衍生物包括对氨基苄氧基羰基(pabc)。本领域技术人员将认识到,自降解基团能够进行化学反应,所述化学反应从有效负载中释放连接体的其余原子。
[0134]
在一些实施方案,所述连接体是:其中:sp1是间隔基团;
sp2是间隔基团;是与所述结合剂连接的一个或多个键;是与所述有效负载连接的一个或多个键;各aa分别为氨基酸残基;和n是从0至10的整数。
[0135]
sp1间隔基团是将(aa)n部分或残基连接至结合剂(ba)或连接至与ba连接的活性基团残基的基团部分。合适的sp1间隔基团包括但不限于,包含亚烷基或聚醚或亚烷基和聚醚两者的那些基团。间隔基团的末端,例如间隔基团连接至所述ba或aa的部分,可以是衍生自活性基团部分的基团部分,所述活性基团部分用于在化学合成偶联物的过程中将抗体或aa偶联至间隔基团的目的。在某些实施方案,n是0、1、2、3、或4(即,当n是0时,aa不存在)。在特定实施方案,n是2。在特定实施方案,n是3。在特定实施方案,n是4。
[0136]
在一些实施方案,所述sp1间隔基团包含亚烷基。在一些实施方案,所述sp1间隔基团包含c
5-7
亚烷基。在一些实施方案,所述sp1间隔基团包含聚醚。在一些实施方案,所述sp1间隔基团包含乙撑氧的聚合物,例如聚乙二醇(peg)。聚乙二醇的聚合单元通常表示为
–
(och2ch2)
p
–
,其中p可以是从1至100的整数。譬如,
–
(och2ch2)2–
亦可表示为
–
och2ch2–
och2ch2–
或peg2。在某些实施方案,所述聚乙二醇是peg1。在某些实施方案,所述聚乙二醇是peg2。在某些实施方案,所述聚乙二醇是peg3。在某些实施方案,所述聚乙二醇是peg4。在某些实施方案,所述聚乙二醇是peg5。在某些实施方案,所述聚乙二醇是peg6。在某些实施方案,所述聚乙二醇是peg7。在某些实施方案,所述聚乙二醇是peg8。在某些实施方案,所述聚乙二醇是peg9。在某些实施方案,所述聚乙二醇是peg
10
。在某些实施方案,所述聚乙二醇是peg
11
。在某些实施方案,所述聚乙二醇是peg
12
。在某些实施方案,所述聚乙二醇是peg
13
。在某些实施方案,所述聚乙二醇是peg
14
。在某些实施方案,所述聚乙二醇是peg
15
。在某些实施方案,所述聚乙二醇是peg
16
。在某些实施方案,所述聚乙二醇是peg
17
。在某些实施方案,所述聚乙二醇是peg
18
。在某些实施方案,所述聚乙二醇是peg
19
。在某些实施方案,所述聚乙二醇是peg
20
。在某些实施方案,所述聚乙二醇是peg
21
。在某些实施方案,所述聚乙二醇是peg
22
。在某些实施方案,所述聚乙二醇是peg
23
。在某些实施方案,所述聚乙二醇是peg
24
。在某些实施方案,所述聚乙二醇是peg
25
。在某些实施方案,所述聚乙二醇是peg
26
。在某些实施方案,所述聚乙二醇是peg
27
。在某些实施方案,所述聚乙二醇是peg
28
。在某些实施方案,所述聚乙二醇是peg
29
。在某些实施方案,所述聚乙二醇是peg
30
。在某些实施方案,所述聚乙二醇是peg
31
。在某些实施方案,所述聚乙二醇是peg
32
。在某些实施方案,所述聚乙二醇是peg
33
。在某些实施方案,所述聚乙二醇是peg
34
。在某些实施方案,所述聚乙二醇是peg
35
。在某些实施方案,所述聚乙二醇是peg
36
。在某些实施方案,所述聚乙二醇是peg
37
。在某些实施方案,所述聚乙二醇是peg
38
。在某些实施方案,所述聚乙二醇是peg
39
。在某些实施方案,所述聚乙二醇是peg
40
。在某些实施方案,所述聚乙二醇是peg
41
。在某些实施方案,所述聚乙二醇是peg
42
。在某些实施方案,所述聚乙二醇是peg
43
。在某些实施方案,所述聚乙二醇是peg
44
。在某些实施方案,所述聚乙二醇是peg
45
。在某些实施方案,所述聚乙二醇是peg
46
。在某些实施方案,所述聚乙二醇是peg
47
。在某些实施方案,所述聚乙二醇是peg
48
。在某些实施方案,所述聚乙二醇是peg
49
。在某些实施方案,所述聚乙二醇是peg
50
。在某些实施方案,所
述聚乙二醇是peg
51
。在某些实施方案,所述聚乙二醇是peg
52
。在某些实施方案,所述聚乙二醇是peg
53
。在某些实施方案,所述聚乙二醇是peg
54
。在某些实施方案,所述聚乙二醇是peg
55
。在某些实施方案,所述聚乙二醇是peg
56
。在某些实施方案,所述聚乙二醇是peg
57
。在某些实施方案,所述聚乙二醇是peg
58
。在某些实施方案,所述聚乙二醇是peg
59
。在某些实施方案,所述聚乙二醇是peg
60
。在某些实施方案,所述聚乙二醇是peg
61
。在某些实施方案,所述聚乙二醇是peg
62
。在某些实施方案,所述聚乙二醇是peg
63
。在某些实施方案,所述聚乙二醇是peg
64
。在某些实施方案,所述聚乙二醇是peg
65
。在某些实施方案,所述聚乙二醇是peg
66
。在某些实施方案,所述聚乙二醇是peg
67
。在某些实施方案,所述聚乙二醇是peg
68
。在某些实施方案,所述聚乙二醇是peg
69
。在某些实施方案,所述聚乙二醇是peg
70
。在某些实施方案,所述聚乙二醇是peg
71
。在某些实施方案,所述聚乙二醇是peg
72
。在某些实施方案,所述聚乙二醇是peg
73
。在某些实施方案,所述聚乙二醇是peg
74
。在某些实施方案,所述聚乙二醇是peg
75
。在某些实施方案,所述聚乙二醇是peg
76
。在某些实施方案,所述聚乙二醇是peg
77
。在某些实施方案,所述聚乙二醇是peg
78
。在某些实施方案,所述聚乙二醇是peg
79
。在某些实施方案,所述聚乙二醇是peg
80
。在某些实施方案,所述聚乙二醇是peg
81
。在某些实施方案,所述聚乙二醇是peg
82
。在某些实施方案,所述聚乙二醇是peg
83
。在某些实施方案,所述聚乙二醇是peg
84
。在某些实施方案,所述聚乙二醇是peg
85
。在某些实施方案,所述聚乙二醇是peg
86
。在某些实施方案,所述聚乙二醇是peg
87
。在某些实施方案,所述聚乙二醇是peg
88
。在某些实施方案,所述聚乙二醇是peg
89
。在某些实施方案,所述聚乙二醇是peg
90
。在某些实施方案,所述聚乙二醇是peg
91
。在某些实施方案,所述聚乙二醇是peg
92
。
[0137]
在一些实施方案,所述sp1间隔基团是:其中:rg
′
是活性基团rg与结合剂反应后的活性基团残基;是与所述结合剂连接的键;是与(aa)n连接的键;n是从0至10的整数;和b独立地为从1至92的整数。
[0138]
活性基团rg可以是本领域技术人员已知的能够与结合剂连接形成一个或多个键的任何活性基团。活性基团rg是在其结构中包含能够与结合剂反应(例如,在抗体的半胱氨酸或赖氨酸残基或叠氮化物基团部分处与抗体反应,例如在一个或多个谷氨酰胺残基处与peg-n3官能化抗体反应;或在氨基基团部分处,例如,在一个或多个谷氨酰胺残基处与peg-nh2官能化抗体反应)以形成本发明所述抗体-药物偶联物的部分的基团部分。与结合剂偶联后,活性基团成为活性基团残基(rg
′
)。示例性活性基团包括但不限于,包含能够与结合剂反应的卤代乙酰基、异硫氰酸酯、琥珀酰亚胺、n-羟基琥珀酰亚胺、或马来酰亚胺部分的那些活性基团。
[0139]
所述sp2间隔基团,当存在时,是将(aa)n部分连接至有效负载的基团部分。合适的
间隔基团包括但不限于以上描述为sp1间隔基团的那些。其他合适的sp2间隔基团包括但不限于包含亚烷基或聚醚或亚烷基和聚醚两者的那些。sp2间隔基团的末端(例如,直接连接至有效负载或aa的间隔基团的部分)可以是衍生自活性基团部分的基团部分,所述活性基团部分用于在化学合成偶联物的过程中将有效负载或aa偶联至sp2间隔基团的目的。在一些实施例,sp2间隔基团的末端(例如,直接连接至有效负载或aa的sp2间隔基团的部分)可以是活性基团部分的残基,所述活性基团部分用于在化学合成偶联物的过程中将有效负载或aa偶联至间隔基团的目的。
[0140]
在一些实施方案,所述sp2间隔基团,当存在时,选自由以下组成的组:
–
nh-(p-c6h4)-ch2–
、
–
nh-(p-c6h4)-ch2oc(o)
–
、氨基酸、二肽、三肽、寡肽、、氨基酸、二肽、三肽、寡肽、及其任何组合。在某些实施方案,各分别为与所述有效负载连接的键,和各分别为与(aa)n连接的键,或者如果n=0,则(aa)n不存在。
[0141]
在上述通式中,各(aa)n分别为氨基酸,或任选地为对氨基苄氧基羰基残基(pabc)。n可以是0;如果是,则(aa)n不存在。如果存在pabc,优选地仅存在一个pabc。优选地,所述pabc残基,若存在,则与(aa)n基团中靠近所述有效负载的末端aa连接。每个aa的合适氨基酸包括天然、非天然的、标准、非标准的、蛋白原、非蛋白原的、和l-型或d-型α-氨基酸。在一些实施方案,所述aa包含丙氨酸、缬氨酸、亮氨酸、异亮氨酸、蛋氨酸、色氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、半胱氨酸、酪氨酸、天冬酰胺、谷氨酰胺、天冬氨酸、谷氨酸、赖氨酸、精氨酸、组氨酸、或瓜氨酸、其衍生物、或其任何组合(例如,二肽类、三肽类、和寡肽类等)。在某些实施方案,所述氨基酸的一个或多个侧链连接至如下所述的侧链基团。在一些实施方案,n是2。在一些实施方案,所述(aa)n是缬氨酸-瓜氨酸。在一些实施方案,(aa)n是瓜氨酸-缬氨酸。在一些实施方案,(aa)n是缬氨酸-丙氨酸。在一些实施方案,(aa)n是丙氨酸-缬氨酸。在一些实施方案,(aa)n是缬氨酸-甘氨酸。在一些实施方案,(aa)n是甘氨酸-缬氨酸。在一些实施方案,所述(aa)n是缬氨酸-瓜氨酸-pabc。在一些实施方案,(aa)n是瓜氨酸-缬氨酸-pabc。在一些实施方案,n是3。在一些实施方案,(aa)n是谷氨酸-缬氨酸-瓜氨酸。在一些实施方案,(aa)n是谷氨酰胺-缬氨酸-瓜氨酸。在一些实施方案,(aa)n是赖氨酸-缬氨酸-丙氨酸。在一些实施方案,(aa)n是赖氨酸-缬氨酸-瓜氨酸。在一些实施方案,n是4。在一些实施方案,(aa)n是谷氨酸-缬氨酸-瓜氨酸-pab。在一些实施方案,(aa)n是谷氨酰胺-缬氨酸-瓜氨酸-pabc。技术人员将认识到pabc是具有以下结构的对氨基苄氧基羰基的残基:所述pabc残基已被证明有助于在体外和体内切割某些连接体。譬如,在某些实施
方案,在pabc切割时,羧酸酯或羧酸基团部分(即,分别为)与抗病毒化合物或有效负载的其余部分保持完整。在某些实施方案,各分别为与抗病毒化合物(例如,有效负载)的其余部分连接的键。技术人员将认识到pab为对氨基苄基(即,
–
nh-(p-c6h4)-ch2–
或)的二价残基。在某些实施方案,所述pab残基已被证明有助于在体外和体内切割某些连接体。譬如,在某些实施方案,在切割时,醇盐或羟基基团部分(即,分别为)与抗病毒化合物(例如,有效负载)的其余部分保持完整。在某些实施方案,各分别为与抗病毒化合物或有效负载的其余部分连接的键。连接体-有效负载
[0142]
在某些实施方案,连接体-有效负载包括由上述式i和ii中任一个或多个所包含的、与连接体连接的任何特定化合物或有效负载,其中本发明所述的连接体包括与本发明所述的抗体或其抗原结合片段反应的基团部分。在特定实施方案,所述连接体与上述式i和ii中任一个或多个中的羧基或羟基连接。在一个实施方案,所述连接体-有效负载具有以下结构:或其药学上可接受的盐,其中l是连接体,如本发明公开的任一实施方案中所述;和rg是活性基团部分,如本发明公开的任一实施方案中所述。在一个实施方案,所述连接体-有效负载是:
或其药学上可接受的盐,其中sp1和sp2,当存在时,均为如本发明公开的任一实施方案中所述的间隔基团;rg是与本发明公开的任一实施方案中所述的抗体或其抗原结合片段反应的基团部分;各aa分别为如本发明公开的任一实施方案中所述的氨基酸;和n是从1至10的整数。
[0143]
在某些实施方案,本发明提供了选自由以下组成的组的化合物(即,连接体-有效负载):
偶联物/抗体-药物偶联物(adc)
[0144]
本发明提供了与治疗性基团部分(例如,类毒素或抗病毒药物)偶联以治疗流感病毒感染的人抗流感-ha单克隆抗体(即,adc)。只要抗体能够结合其靶标,所述抗体便可在沿着所述抗体的任何位置处与治疗剂连接。在一个实施方案,所述治疗剂可以是针对流感-ha的第二种不同抗体或其adc。在某些实施方案,所述抗体可与病毒感染细胞特异性的药剂偶联。可与抗流感-ha抗体偶联的治疗性基团部分的类型需要考虑待治疗的病症以及要实现的所需治疗效果。在某些实施方案,本发明提供了抗体或其抗原结合片段,其中所述抗体与本发明所述的一种或多种式i和/或式ii所示化合物偶联。在一个实施方案,所述抗流感抗体或其抗原结合片段经由连接体偶联至有效负载,其各自如本发明公开的其各自的实施方案中任一个所述。
[0145]
在一个实施方案,所述抗体-药物偶联物具有以下结构:其中ab是抗流感抗体或其抗原结合片段;l是连接体;和p是抗病毒化合物或有效负载。在一个实施方案,ab是抗流感抗体或其抗原结合片段;和p是流感抑制剂。在一个实施方案,ab是抗流感抗体或其抗原结合片段;和p是聚合酶抑制剂。在一个实施方案,ab是抗流感抗体或其抗原结合片段;和p是vx-787、其衍生物、或其残基。在一个实施方案,ab是抗流感抗体或其抗原结合片段;和p是巴洛沙韦(baloxavir)、其衍生物、或其残基。在一个实施方案,ab是抗血凝素抗体或其抗原结合片段;和p是抗病毒化合物。在一个实施方案,ab是抗流感抗体或其抗原结合片段;和p是抗病毒化合物。在一个实施方案,ab是抗血凝素抗体或其抗原结合片段;和p是流感抑制剂。在一个实施方案,ab是抗血凝素抗体或其抗原结合片段;和p是聚合酶抑制剂。在本段的任一实施方案中,ab是抗流感抗体或其抗原结合片段或抗血凝素抗体或其抗原结合片段,其中所述抗体与上述式i所示化合物偶联。在本段的任一实施方案中,ab是抗流感抗体或其抗原结合片段或抗血凝素抗体或其抗原结合片段,其中所述抗体与上述式ii所示化合物偶联。在一个实施方案,ab是抗血凝素抗体或其抗原结合片段;和p是vx-787、其衍生物、或其残基。在一个实施方案,ab是抗血凝素抗体或其抗原结合片段;和p是巴洛沙韦(baloxavir)、其衍生物、或其残基。在本段的任一实施方案中,k是从1至30的整数。在某些实施方案,本发明提供了adc,其中所述抗体或其抗原结合片段偶联至所示化合物、或其药学上可接受的盐,其中,l是本发明所述的连接体;和rg是本发明所述的活性基团部分。在某些实施方案,本发明提供了adc,其中所述经偶联的化合物选自
其中l是本发明所述的连接体。
[0146]
在某些实施方案,本发明提供了具有以下结构的adc化合物:其中l和ba均如本发明其他地方所述,和k是1、2、3、4、5、6、7、8、9或10。在某些实施方案,k的范围为1-2、1-3、2-3、2-4、3-4、或1-4。在某些实施方案,如上偶联至—l—ba的化合物包括一种或多种上述式i和/或式ii所示化合物,其中ba是结合剂;l是连接体;和k是1、2、3、4、5、6、7、8、9或10。在某些实施方案,其中如上偶联至—l—ba的化合物包括一种或多种上述式i和/或式ii所示化合物,其中ba是结合剂和l是连接体,k的范围为1-2、1-3、2-3、2-4、3-4、或1-4。
[0147]
在一个实施方案,本发明提供了选自由以下组成的组的adc化合物:
其中ba是抗体或其抗原结合片段;和k是从1至30的整数。在某些实施方案,k是1、2、3、4、5、6、7、8、9或10。在某些实施方案,k的范围为1-2、1-3、1-4、1-5、1-6、1-7、1-8、1-9、1-10、2-3、2-4、2-5、2-6、2-7、2-8、2-9、2-10、3-4、3-5、3-6、3-7、3-8、3-9、3-10、4-5、4-6、4-7、4-8、4-9、4-10、5-6、5-7、5-8、5-9、5-10、6-7、6-8、6-9、6-10、7-8、7-9、7-10、8-9、8-10、或9-10。在本段的任一实施方案中,当k大于1时,考虑ba包括一个或多个半胱氨酸、赖氨酸和/或谷氨酰胺残基以用于偶联至有效负载和/或连接体-有效负载。譬如,上述adc描述考虑了一种或多种情况的药物:抗体比值(dar),其中ba的一个或多个半胱氨酸、赖氨酸和/或谷氨酰胺残基容纳有效负载和/或连接体-有效负载(例如,当k是≥1)。ba与
–s–
和/或ba与
–
nh
–
之间的键分别表示结合剂(ba)与ba的半胱氨酸或经转谷氨酰胺酶化的谷氨酰胺残基之间的键。并且,ba
–s–
和/或ba
–
nh
–
与碳之间的键表示与本发明其他地方所示或所述的连接体连接的键。因此,来自ba的半胱氨酸残基的s和/或来自ba的经转谷氨酰胺酶化的谷氨酰胺残基的n均示出在括号内,以表明ba可与一个以上的有效负载和/或连接体-有效负载偶联(例如,ba的dar≥1)。在一个实施方案,ba是如本发明所述的抗体或其抗原结合片段。
[0148]
在本发明所述的某些adc实施方案中,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少一个用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少两个用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少三个用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少四个用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少一个可用于偶联的谷氨酰胺残基。在一个实施方案,ab
或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少两个可用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少三个可用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其包含至少四个可用于偶联的谷氨酰胺残基。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在两个q295残基处进行;和k是2。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在eu编号系统中的两个q295残基处进行;和k是2。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在所述重链的所述c端进行;和k是2。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由谷氨酰胺进行。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由谷氨酰胺进行;和k是2。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由所述抗体重链的所述c端的llqga序列中的谷氨酰胺进行。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由所述抗体重链的所述c端的llqga序列中的谷氨酰胺进行;和k是2。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在两个q295残基和两个n297q残基处进行;和k是4。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在eu编号系统中的两个q295残基以及两个n297q残基处进行;和k是4。在一个实施方案,ab或ba是如本发明所述的mab11729。
[0149]
在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在两个q295残基处进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0;g)约1.0;或h)约2.0。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在eu编号系统中的两个q295残基处进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0;g)约1.0;或h)约2.0。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在所述重链的所述c端进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0;g)约1.0;或h)约2.0。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由谷氨酰胺进行。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由谷氨酰胺进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0;g)约1.0;或h)约2.0。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由所述抗体重链的所述c端的llqga序列中的谷氨酰胺进行。在一个实施方案,ab或ba是包含抗体重链的经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是经由所述抗体重链的所述c端的llqga序列中的谷氨酰胺进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0;g)约1.0;或h)约2.0。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在两个q295残基和两个n297q残基处进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约
0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0或约3.0或约4.0;g)约1.0;h)约2.0;i)约3.0;或j)约4.0。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在eu编号系统中的两个q295残基以及两个n297q残基处进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0;g)约1.0;或h)约2.0。在一个实施方案,ab或ba是经转谷氨酰胺酶修饰的抗体或其抗原结合片段,其中偶联是在两个q295残基和两个n297q残基处进行;和dar是:a)约2.0;b)大于0至约12.0;c)约0.5至约8.0;d)约0.5至约6.0;e)约1.0至约4.0;f)约1.0或约2.0或约3.0或约4.0;g)约1.0;h)约2.0;i)约3.0;或j)约4.0。在一个实施方案,ab或ba是如本发明所述的mab11729。
[0150]
在本发明所述的某些adc实施方案中,ab或ba是抗流感抗体或其抗原结合片段。在一个实施方案,ab或ba是抗甲型流感抗体或其抗原结合片段。在一个实施方案,ab或ba是抗甲型流感组1抗体或其抗原结合片段。在一个实施方案,ab或ba是抗流感h1抗体或其抗原结合片段。在一个实施方案,ab或ba是抗甲型流感组2抗体或其抗原结合片段。在一个实施方案,ab或ba是抗流感h3抗体或其抗原结合片段。在一个实施方案,ab或ba是抗乙型流感抗体或其抗原结合片段。在一个实施方案,adc包含经由连接体与有效负载偶联的抗流感抗体或其抗原结合片段,其中所述抗体-药物偶联物结合和/或抑制聚合酶碱性蛋白2(pb2)(vx-787)、聚合酶酸性蛋白(pa)(巴洛沙韦和/或巴洛沙韦酯(baloxavir marboxil))、和/或聚合酶碱性蛋白1(pb1)。在一个实施方案,adc包含经由连接体与有效负载偶联的抗流感抗体或其抗原结合片段,其中所述抗体-药物偶联物结合和/或抑制聚合酶碱性蛋白2(pb2)(vx-787),通过elisa测定的亲和性为至少4.0x10-9
m、至少3.5x10-9
m、或至少3.0x10-9
m。在一个实施方案,adc包含经由连接体与有效负载偶联的抗流感抗体或其抗原结合片段,其中所述抗体-药物偶联物结合和/或抑制聚合酶碱性蛋白1(pb1),通过elisa测定的亲和性为至少4.0x10-9
m、至少3.5x10-9
m、或至少3.0x10-9
m。在一个实施方案,adc包含经由连接体与有效负载偶联的抗流感抗体或其抗原结合片段,其中所述抗体-药物偶联物结合和/或抑制聚合酶碱性蛋白2(pb2)(vx-787),通过分析测定的ic
50
为至少2.5x10-9
m、至少2.0x10-9
m、或至少1.5x10-9
m。在一个实施方案,adc包含经由连接体与有效负载偶联的抗流感抗体或其抗原结合片段,其中所述抗体-药物偶联物结合和/或抑制聚合酶碱性蛋白1(pb1),通过分析测定的ic
50
为至少2.5x10-9
m、至少2.0x10-9
m、或至少1.5x10-9
m。制备化合物或有效负载以及连接体-有效负载的方法
[0151]
本发明提供的化合物可通过本领域技术人员显而易见的任何方法制备、分离或得到。示例性制备方法在下文实施例中详细描述。在某些实施方案,本发明提供的化合物是可商购得到或通常可根据方案a-c制备得到:方案a.示例性制备方案
方案b1.示例性制备方案
方案b2.示例性制备方案方案b3.示例性制备方案
[0152]
在上述示例性制备方案a(参见,j.med.chem.2014,57,6668)中,r1如式i上下文中所述。在方案a中,在与马来酸酐和1,3-环己二烯的狄-阿(diels-alder)环加成反应之后,可在碱性条件下搅拌內型-a1以提供差向异构化的反式-a2。curtius重排并用苯甲醇捕获以提供a3。氢化提供a4。用2,4-二氯嘧啶类化合物处理并手性分离,经由中间体a5提供a6。与取代的氮杂吲哚硼酸酯类化合物进行suzuki偶联,然后脱保护,生成式i所示化合物,包括vx-787及其衍生物。
[0153]
在上述示例性制备方案b1-b3(参见,oprd 2019,23,1298)中,r1如式ii上下文中所述。在方案b1中,可对b1进行保护以提供b2。然后将b2还原以提供b3,然后用羟基取代甲氧基以提供b4。b5可进行酯化以提供b6。b6可用boc-肼处理得到吡啶酮b7,然后在酸性条件下脱保护得到b8。在路易斯酸存在下将b4和b8组合以提供二取代的肼b9。氮脱保护以及pd介导的环化提供了内消旋-b10(rac-b10)。通过形成酰肼非对映异构体来分离内消旋-b10(rac-b10),然后水解生成b12。
[0154]
在方案b2中,将b13进行邻位金属化并用dmf淬灭以提供醛,所述醛与羧酸进行分子内环化以提供b14。b14可用苯硫酚处理以提供b15。将b15还原以提供b16。通过在酸性条件下处理b16,提供三环硫化物b17。将b17还原以提供b18。
[0155]
在方案b3中,将b12与b18组合以提供巴洛沙韦及其衍生物。脱保护以提供巴洛沙韦酸b19。巴洛沙韦酸b19可再进行烷基化以提供b20。
[0156]
本发明所述的连接体-有效负载通常可通过一系列偶联步骤合成得到,如方案c所示:方案c.示例性制备方案
[0157]
在上述示例性制备方案c中,r1如式i上下文中所述。在方案c中,vx-787及其衍生物用带有离去基团(lg)的连接体处理以提供连接体-有效负载(例如,连接体-(vx-787))。
[0158]
本发明所述的偶联物可通过将本发明所述的连接体-有效负载与结合剂(例如,本发明所述的抗体)在标准偶联条件下进行偶联合成得到(参见例如,doronina et al.nature biotechnology 2003,21,7,778,其通过引用其全文并入本发明)。当结合剂是抗体时,所述抗体可经由所述抗体的一个或多个半胱氨酸或赖氨酸残基与连接体-有效负载偶联。连接体-有效负载可以,例如通过使抗体经受还原剂(例如,二硫苏糖醇)的作用以切割所述抗体的二硫键,例如通过凝胶过滤纯化所述经还原的抗体,然后用含有合适的活性基团部分(例如,马来酰亚胺基基团)的连接体-有效负载处理所述抗体,从而与半胱氨酸残基偶联(参见例如,示例性制备方案c)。合适的溶剂包括但不限于水、dma、dmf、和dmso。含有活性基团例如活化的酯或酰基卤基团的连接体-有效负载可与抗体的赖氨酸残基偶联。合适的溶剂包括但不限于水、dma、dmf、和dmso。偶联物可使用已知的蛋白质技术进行纯化,包括例如体积排除色谱法(尺寸排阻色谱法)、透析法和超滤/渗滤法。
[0159]
结合剂,例如抗体,也可通过点击化学反应进行偶联。在所述点击化学反应的一些实施方案中,连接体-有效负载包括能够与叠氮化物进行区域异构的1,3-环加成反应的活
性基团,例如炔。此类合适的活性基团如上所述。所述抗体包括一个或多个叠氮基团。此类抗体包括用例如叠氮基-聚乙二醇基团官能化的抗体。在某些实施方案,此类官能化抗体通过在酶转谷氨酰胺酶存在下,用伯胺化合物处理具有至少一个谷氨酰胺残基(例如,重链gln295)的抗体,进行衍生得到。在某些实施方案,此类官能化抗体通过在酶转谷氨酰胺酶存在下,用伯胺化合物处理具有至少一个谷氨酰胺残基(例如,重链gln297)的抗体,进行衍生得到。此类抗体包括asn297gln(n297q)突变体。在某些实施方案,此类官能化抗体通过在酶转谷氨酰胺酶存在下,用伯胺化合物处理具有至少两个谷氨酰胺残基(例如,重链gln295和重链gln297)的抗体,进行衍生得到。此类抗体包括asn297gln(n297q)突变体。在某些实施方案,所述抗体具有如本段所述的两条重链,用于总共两个或总共四个谷氨酰胺残基。
[0160]
在某些实施方案,此类官能化抗体是通过在酶转谷氨酰胺酶存在下,用伯胺化合物或肽标签处理具有至少一个谷氨酰胺残基(例如,重链gln295)的抗体,进行衍生得到。在某些实施方案,此类官能化抗体是通过在酶转谷氨酰胺酶存在下,用伯胺化合物或肽标签处理具有至少一个谷氨酰胺残基(例如,重链gln297)的抗体,进行衍生得到。此类抗体包括asn297gln(n297q)突变体。在某些实施方案,此类官能化抗体是通过在酶转谷氨酰胺酶存在下,用伯胺化合物或肽标签处理具有至少两个谷氨酰胺残基(例如,重链gln295和重链gln297)的抗体,进行衍生得到。此类抗体包括asn297gln(n297q)突变体。在某些实施方案,所述抗体具有两条如本段所述的重链,用于总共两个或总共四个谷氨酰胺残基。
[0161]
在一个实施方案,所述官能化抗体或抗原结合分子包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签。在一个实施方案,所述官能化抗体或抗原结合分子包括抗体重链并且进一步包括位于所述抗体重链的所述c端的肽标签,其中所述肽标签是五肽序列llqga。在实施方案中,所述官能化抗体或抗原结合分子包括两条抗体重链并且进一步包括位于每条抗体重链的所述c端的肽标签。在一个实施方案,所述官能化抗体或抗原结合分子包括两条抗体重链并且进一步包括位于每条抗体重链的所述c端的肽标签,其中所述肽标签是五肽序列llqga。
[0162]
在某些实施方案,所述抗体包含两个谷氨酰胺残基,每条重链中有一个。在特定实施方案,所述抗体在每条重链中包含q295残基。在进一步的实施方案,所述抗体包含1、2、3、4、5、6、7、8个、或更多个谷氨酰胺残基。所述这些谷氨酰胺残基可位于重链中、轻链中、或位于重链和轻链两者中。所述这些谷氨酰胺残基可以是野生型残基、或工程化残基。所述抗体可根据标准技术制备得到。
[0163]
本领域技术人员将认识到,抗体通常在重链序列中的残基q295附近的残基n297处进行糖基化。残基n297处的糖基化可干扰残基q295处的转谷氨酰胺酶(dennler et al.,同上)。因此,在有利的实施方案中,所述抗体未被糖基化。在某些实施方案,所述抗体是去糖基化的或无糖基化的。在特定实施方案,抗体重链具有n297突变。换言之,所述抗体突变为在位置297处不再具有天冬酰胺残基。在特定实施方案,抗体重链具有n297q突变。此类抗体可通过定点诱变除去糖基化序列或使糖基化序列失效或通过定点诱变以在任何干扰糖基化位点或任何其他干扰结构之外的位点处插入谷氨酰胺残基,来制备得到。亦可从天然来源或人工来源分离得到此类抗体。
[0164]
然后使不干扰糖基化的抗体与伯胺化合物反应或用伯胺化合物处理。在某些实施方案,使无糖基化的抗体与伯胺化合物反应或用伯胺化合物处理以生成经谷氨酰胺酰基修
饰的抗体。在某些实施方案,使去糖基化的抗体与伯胺化合物反应或用伯胺化合物处理以生成经谷氨酰胺酰基修饰的抗体。
[0165]
伯胺可以是在转谷氨酰胺酶存在下能够与谷氨酰胺残基形成共价键的任何伯胺。有用的伯胺如本发明所述(参见例如,示例性制备方案c)。所述转谷氨酰胺酶可以是本领域技术人员认为合适的任何转谷氨酰胺酶。在某些实施方案,所述转谷氨酰胺酶是催化伯胺化合物上的游离胺基团与谷氨酰胺残基侧链上的酰基基团之间的异肽键形成的酶。转谷氨酰胺酶亦称为蛋白质-谷氨酰胺-γ-谷氨酰转移酶。在特定实施方案,所述转谷氨酰胺酶被分类为ec 2.3.2.13。所述转谷氨酰胺酶可来自认为合适的任何来源。在某些实施方案,所述转谷氨酰胺酶是微生物。有用的转谷氨酰胺酶已从茂原链轮丝菌(streptomyces mobaraense)、肉桂链霉菌(streptomyces cinnamoneum)、灰肉链霉菌(streptomyces griseo-carneum)、淡紫灰链霉菌(streptomyces lavendulae)和枯草芽孢杆菌(bacillus subtilis)中分离得到。也可使用非微生物的转谷氨酰胺酶,包括哺乳动物的转谷氨酰胺酶,例如,非微生物转谷氨酰胺酶与辅因子组合。在某些实施方案,所述转谷氨酰胺酶可通过任何技术生成或从本领域技术人员认为合适的任何来源得到。在特定实施方案,所述转谷氨酰胺酶得自商业来源。
[0166]
在某些实施方案,将经谷氨酰胺酰基修饰的抗体与活性连接体-有效负载反应或用活性连接体-有效负载处理以形成抗体-连接体-有效负载偶联物。所述反应可在本领域技术人员认为合适的条件下进行。在某些实施方案,使经谷氨酰胺酰基修饰的抗体与活性连接体-有效负载化合物在适于在经谷氨酰胺酰基修饰的抗体与连接体-有效负载化合物之间形成键的条件下接触。合适的反应条件是本领域技术人员周知的。药物组合物和治疗方法
[0167]
本发明提供了治疗和预防疾病、病症或病况的方法,其包含施用治疗或预防有效量的一种或多种本发明公开的化合物,例如一种或多种本发明所提供通式的化合物。疾病、病症和/或病况包括但不限于与本发明所述的病毒感染相关的那些。
[0168]
本发明所述的化合物可以单独施用或与一种或多种附加(补充)治疗剂一同施用。可在施用本发明所述化合物之前、同时或之后不久施用一种或多种附加治疗剂。本发明还包括药物组合物,其包含任何本发明所述化合物与一种或多种附加治疗剂组合,以及治疗方法,所述方法包含向有需要的受试者施用此类组合。
[0169]
合适的附加治疗剂包括但不限于:抗病毒药物(例如,第二抗病毒化合物或有效负载)、自身免疫治疗剂、激素、生物制品、或单克隆抗体。在一些实施方案,所述补充治疗剂可选自由以下组成的组:抗病毒药物、抗炎药(例如,皮质类固醇或非甾体抗炎药)、特异性结合流感ha的抗体、流感疫苗、膳食补充剂(例如,抗氧化剂)、和用于治疗流感感染的姑息疗法。在一个实施方案,所述抗炎药选自由皮质类固醇类和非甾体抗炎药类组成的组。在一个实施方案,所述膳食补充剂是抗氧化剂。合适的治疗剂还包括但不限于,本发明所述的抗病毒化合物或有效负载的任何药学上可接受的盐类或衍生物类。在一些实施方案,所述补充治疗剂通过与本发明所述的抗体-药物偶联物、化合物或药物组合物不同的给药途径进行施用。例如,补充治疗剂可进行口服施用。作为补充治疗剂施用的示例性抗病毒药物是奥司他韦(oseltamivir)。在一些实施方案,在施用所述抗体-药物偶联物、化合物或药物组合物之前施用奥司他韦。在一些实施方案,奥司他韦与所述抗体-药物偶联物、化合物或药物组
合物同时施用。在一些实施方案,在施用所述抗体-药物偶联物、化合物或药物组合物之后施用奥司他韦。在一些实施方案,所述抗病毒药物是抗甲型流感药物或抗乙型流感药物(例如,抗体或其抗原结合部分),例如特异性结合甲型流感ha的抗体或特异性结合乙型流感ha的抗体。
[0170]
在本发明所述方法的一些实施方案中,可在限定的时间疗程内向受试者施用多剂量的本发明所述化合物(或药物组合物,其包含本发明所述化合物与本发明提及的任何附加治疗剂的组合)。根据本发明内容的此实施方案的方法包括向受试者依次施用多剂量的本发明所述化合物。本发明包含以下方法,所述方法包含依次向患者施用单次初始剂量的本发明所述化合物,随后施用一次或多次二次剂量的所述化合物,以及任选地后续施用一次或多次三次剂量的所述化合物。本发明化合物的示例性剂量包括但不限于50mg/kg、49mg/kg、48mg/kg、47mg/kg、46mg/kg、45mg/kg、44mg/kg、43mg/kg、42mg/kg、41mg/kg、40mg/kg、39mg/kg、38mg/kg、37mg/kg、36mg/kg、35mg/kg、34mg/kg、33mg/kg、32mg/kg、31mg/kg、30mg/kg、29mg/kg、28mg/kg、27mg/kg、26mg/kg、25mg/kg、24mg/kg、23mg/kg、22mg/kg、21mg/kg、20mg/kg、19mg/kg、18mg/kg、17mg/kg、16mg/kg、15mg/kg、14mg/kg、13mg/kg、12mg/kg、11mg/kg、10mg/kg、9mg/kg、8mg/kg、7mg/kg、6mg/kg、5mg/kg、4mg/kg、3mg/kg、2mg/kg、1mg/kg、0.9mg/kg、0.8mg/kg、0.7mg/kg、0.6mg/kg、0.5mg/kg、0.4mg/kg、0.3mg/kg、0.2mg/kg、0.1mg/kg、和0.05mg/kg。
[0171]
在某些实施方案,在治疗过程中,所述初始、二次和/或三次剂量中所含的化合物的量彼此变化(例如,酌情上调或下调)。在某些实施方案,在治疗方案开始时,将两种或更多种(例如,2、3、4或5种)剂量施用为“负荷剂量”,随后以较不频繁的方式施用后续剂量(例如“维持剂量”)。
[0172]
在本发明所述的某些示例性实施方案中,每个二次和/或三次剂量在紧随前述剂量之后进行给药1至26周(例如,1、1 1/2、2、2 1/2、3、3 1/2、4、4 1/2、5、5 1/2、6、6 1/2、7、7 1/2、8、8 1/2、9、9 1/2、10、10 1/2、11、11 1/2、12、12 1/2、13、13 1/2、14、14 1/2、15、15 1/2、16、16 1/2、17、17 1/2、18、18 1/2、19、19 1/2、20、20 1/2、21、21 1/2、22、22 1/2、23、23 1/2、24、24 1/2、25、25 1/2、26、26 1/2、或更多)。
[0173]
本发明此实施方案的方法可包含向患者施用任何数量的二次和/或三次剂量的本发明化合物。譬如,在某些实施方案,仅向患者施用单次二次剂量。在其他实施方案,向患者施用两次或更多次(例如,2、3、4、5、6、7、8次或更多次)二次剂量。同理,在某些实施方案,仅向患者施用单次三次剂量。在其他实施方案,向患者施用两次或更多次(例如,2、3、4、5、6、7、8次或更多次)三次剂量。给药方案可在特定受试者的寿命期内无限期地进行,或者直至此种治疗不再是治疗需要或有利的。
[0174]
在涉及多个二次剂量的实施方案中,每个二次剂量可以与其他二次剂量相同的频率施用/给药。譬如,每个二次剂量可在紧随前述剂量之后,对患者进行给药1至2周或1至2个月。同理,在涉及多个三次剂量的实施方案中,每个三次剂量可以与其他三次剂量相同的频率施用/给药。譬如,每个三次剂量可在紧随前述剂量之后,对患者进行给药2至12周。在本发明的某些实施方案中,向患者施用二次和/或三次剂量的频率可在治疗方案的过程中变化。也可根据临床检查后的个体患者的需要,由医师在治疗过程中调整给药频率。
[0175]
本发明包括其中以第一频率(例如,每周一次,每两周一次,每三周一次,每月一
次,每两个月一次等)向患者施用2至6次负荷剂量,然后以较不频繁的方式向患者施用两次或更多次维持剂量的给药方案。例如,根据本发明的此实施方案,如果以每月一次的频率施用负荷剂量,那么维持剂量可以每6周施用一次,每两个月施用一次,每三个月施用一次等。
[0176]
本发明包括本发明所述化合物、和/或偶联物(例如,式i和ii所示化合物的抗体-药物偶联物)的药物组合物,譬如,组合物包含本发明所述化合物、其盐、立体异构体、区域异构体、多晶型物,以及药学上可接受的载体、稀释剂、和/或辅料。合适的载体、稀释剂和辅料的实例包括,但不限于:用于维持适当组合物ph值的缓冲剂类(例如,柠檬酸盐缓冲液、琥珀酸盐缓冲液、乙酸盐缓冲液、磷酸盐缓冲液、乳酸盐缓冲液、草酸盐缓冲液等),载体蛋白质类(例如,人血清白蛋白),盐水,多元醇类(例如,海藻糖、蔗糖、木糖醇、山梨糖醇等),表面活性剂类(例如,聚山梨醇酯20、聚山梨醇酯80、聚氧乙烯酸酯(polyoxolate)等),抗微生物剂类,和抗氧化剂类。
[0177]
在某些实施方案,所述化合物或有效负载、连接体-有效负载、adc、或其组合物可通过替代的给药途径进行提供。在某些实施方案,所述组合物的给药途径选自由以下组成的组:皮下、皮内、肌内、口服、静脉内、腹腔内、吸入、和鼻内。在一个实施方案,所述组合物的给药途径是口服。在一个实施方案,所述组合物的给药途径是静脉内。在一个实施方案,所述组合物的给药途径是腹腔内。在一个实施方案,所述组合物的给药途径是吸入。在一个实施方案,所述组合物的给药途径是鼻内。
[0178]
在一些实施例,本发明阐述了治疗、预防、减少或抑制受试者与感染相关的疾病、病症或病况的方法,其包含向所述受试者施用有效量或治疗有效量的式i和/或ii所示化合物、本发明所述的连接体-有效负载、和/或本发明所述的adc、其组合、或其药物组合物。在一些实施方案,所述感染是病毒感染。在一些实施方案,所述感染是流感病毒感染。在一些实施方案,所述感染是甲型流感病毒感染。在一些实施方案,所述感染是乙型流感病毒感染。在一些实施方案,所述感染是甲型流感病毒感染和乙型流感病毒感染。在某些实施方案,与将经偶联的有效负载或adc施用于可比受试者相比,与向所述受试者施用未偶联的有效负载相关的副作用降低。
[0179]
本发明公开的化合物还可用于治疗、预防、减少或抑制受试者的流感感染,包含向所述受试者施用有效量的本发明所述的抗体-药物偶联物、化合物、或药物组合物。在一些实施方案,所述流感感染是由甲型流感病毒感染引起。在一些实施方案,所述流感感染是由甲型流感组1病毒引起。在一些实施方案,所述流感感染是由甲型h1流感病毒引起。在一些实施方案,所述流感感染是由甲型流感组2病毒引起。在一些实施方案,所述流感感染是由甲型h3流感病毒引起。在一些实施方案,所述流感感染是由未知或未确定的流感病毒引起。在一些实施方案,所述流感感染是由乙型流感病毒感染引起。在一些实施方案,所述流感感染是由甲型流感病毒感染和乙型流感病毒感染引起。在一些实施方案,所述流感感染是由甲型流感病毒感染、甲型流感组1病毒感染、甲型h1流感病毒感染、甲型流感组2病毒感染、甲型h3流感病毒感染、未知或未确定的流感病毒、乙型流感病毒感染、或其任何组合引起。实施例
[0180]
本发明提供了vx-787及其衍生物、巴洛沙韦(baloxavir)及其衍生物、巴洛沙韦酯(baloxavir marboxil)及其衍生物、其蛋白质偶联物,以及用于治疗疾病、病症和病况的方法,其包括施用vx-787、巴洛沙韦(baloxavir)和巴洛沙韦酯(baloxavir marboxil)及其偶
联物。实施例1:连接体-有效负载合成
[0181]
vx-787和巴洛沙韦(baloxavir)均用作通过抗血凝素抗体递送的有效负载。为了测试不同连接体与vx-787或巴洛沙韦组合的效果,如下所示合成连接体-有效负载:连接体-(vx-787):化合物6和化合物11;连接体-巴洛沙韦:化合物15。所使用的全部溶剂均按原样使用并从sigma aldrich或fisher scientific购得。1h光谱在varian inova 300mhz和500mhz nmr仪器上记录。相对于用于分析的nmr溶剂,化学位移(δ)以ppm为单位报告,并报告为s
–
单峰、d
–
双峰、t
–
三重峰、q
–
四重峰、dd
–
双二重峰、dt
–
双三重峰、dq
–
双四重峰、和m
–
多重峰。耦合常数(j)以赫兹(hz)为单位报告。使用fastgradient rp-18e分析柱(50
×
2mm,merck kgaa,p/n 1.52007.0001)在配备6130四极杆lc/ms或1200系列lc/ms系统的agilent 1100、1260infinity上,采用以下分析型hplc方法测定色谱纯度:进样量2-10l;流速1ml/min;5-95%乙腈水溶液,历时4分钟;agilent二极管阵列检测器,λ=254nm;室温。使用电喷雾电离源在agilent系统上进行低分辨率质谱分析,并使用单四极杆或离子阱质量检测器进行分析。
[0182]
化合物6由vx-787合成得到,如下文方案1中所述。方案1
[0183]
化合物2:化合物2采用pct国际申请号2014145090中所示方法制备得到。((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代-5-脲基戊-2-基)氨基)-3-甲基-1-氧代丁-2-基)氨基甲酸叔丁酯1(700mg,1.46mmol)溶于ch3cn/h2o/tfa(3:1:1=v/v/v,6ml/2ml/2ml)的混合物。将反应混合物在室温下搅拌19小时,通过lcms进行监测。真空浓缩后,粗产物2(0.5g盐)不经进一步纯化即可直接用于下一步骤。ms(esi,pos.):计算值c
18h29
n5o4,379.2;实测值380.2(m h)。
[0184]
化合物4:将化合物2(100mg,0.263mmol)、6-(fmoc-氨基)己酸3(93mg,
0.263mmol)、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化六氟磷酸盐(hatu,200mg,0.526mmol)和1-羟基-7-氮杂苯并三唑(hoat,35mg,0.263mmol)装入烘干的2打兰(dram)小瓶。然后加入无水dmf(2ml),将反应混合物在环境温度下老化5分钟,然后通过注射器逐滴加入n,n-二异丙基乙胺(diea,137μl,0.789mmol)。将均匀的黄色溶液在室温和氮气下搅拌2小时,通过lc/ms监测反应完成。将反应混合物在50g c18 aq gold色谱柱上纯化(梯度洗脱:10-95%mecn/水,mecn和水两者均含0.05%乙酸),持续20分钟。合并纯级分,在干冰上冷冻,冻干,得到标题化合物4,为白色固体(120mg,65%)。ms:计算值c
39h50
n6o7,714.3;实测值715.3(m h),737.3(m na)。
[0185]
化合物5:在室温下,将化合物4(39.4mg,0.055mmol)和4-二甲氨基吡啶(dmap,5mg,0.039mmol)加至vx-787(22mg,0.055mmol)的无水thf(6ml)于氩气下搅拌的悬浮液中。然后将n,n
’‑
二环己基碳二亚胺(dcc,17mg,0.083mmol)的无水thf(2ml)溶液逐滴加至反应混合物中。搅拌16小时后,混合物蒸发至干,将残余物溶于3ml dmso。粗物料在50g c18 aq gold色谱柱上纯化(梯度洗脱:10-95%mecn/水,mecn和水两者均含0.05%acoh)。合并产物级分,在干冰上冷冻,冻干,得到标题化合物5,为白色固体(38mg,63%)。ms(esi,pos.):计算值c
59h67
f2n
11
o8,1095.5;实测值1096.4(m h)。
[0186]
化合物6:将5%哌啶(0.8ml)的dmf溶液加至在氩气、环境温度下搅拌30分钟的化合物5(33mg,0.0301mmol)的n,n-二甲基甲酰胺(dmf,1ml)溶液中,搅拌所得溶液。lc/ms确证反应完成。反应物于30g c18 aq.gold色谱柱上通过isco系统直接纯化(梯度洗脱:10-95%mecn/水,mecn和水两者均含0.05%乙酸,洗脱30分钟)。合并含有产物的级分,在干冰上冷冻,冻干过夜,得到标题化合物6,为灰白色固体(22mg,85%)。ms:计算值c
44h57
f2n
11
o6,873.4;实测值874.4(m h)。1h-nmr(500mhz;dmso-d6):δ9.97(s,1h),8.50(dd,j=9.8,2.8hz,1h),8.28(dd,j=2.5,1.2hz,1h),8.20(s,1h),8.16(d,j=3.9hz,1h),8.11-8.09(m,1h),7.84(dd,j=1.6,0.9hz,1h),7.62(d,j=6.9hz,1h),7.54(d,j=8.5hz,2h),7.23(d,j=8.5hz,2h),6.02-6.00(m,1h),5.43(d,j=0.3hz,2h),5.09(d,j=12.5hz,1h),5.00(d,j=12.5hz,1h),4.77-4.74(m,1h),4.38-4.36(m,1h),4.19(dd,j=8.5,7.2hz,1h),3.03-2.94(m,4h),2.17(d,j=11.5hz,2h),1.99-1.95(m,3h),1.85(s,2h),1.81-1.78(m,2h),1.73-1.71(m,4h),1.50-1.47(m,8h),1.39-1.34(m,4h),1.26-1.23(m,2h),0.85(dd,j=12.7,6.8hz,6h)。
[0187]
化合物11由vx-787合成得到,如下文方案2所述。方案2
[0188]
化合物8:在室温下,将n-乙氧基羰基-2-乙氧基-1,2-二氢喹啉(eedq,1.99g,8.05mmol)加至对氨基苯甲醇(0.99g,8.05mmol)的二氯甲烷(19ml)和甲醇(7.6ml)溶液中。搅拌5分钟后,一次性加入fmoc-缬氨酸-瓜氨酸7(2.0g,4.03mmol),将所得溶液搅拌18小时。真空除去挥发物,残余物用乙醚(20ml)研磨,依次用乙醚(20ml)、乙酸乙酯(20ml)和乙醚(20ml)洗,得到标题化合物8(2.2g,98%收率)为淡黄色固体。ms(esi,pos.):计算值c
33h39
n5o6,601.29;实测值602.3(m h)。
[0189]
化合物2:在20ml小瓶中,将fmoc-缬氨酸-瓜氨酸-pab(oh)8(2.0g,3.33mmol)溶于5%哌啶的dmf(10ml)溶液并在室温下搅拌1小时。通过过滤除去沉淀物,滤液在100g c18 aq色谱柱上使用梯度洗脱5-95%mecn/h2o(两者均含0.05%tfa)进行纯化。合并纯级分,冷冻,冻干。对冻干的固体再次纯化,得到标题化合物2(0.98g,61%收率),为蓬松的灰白色固体。ms(esi,pos.):计算值c
18h29
n5o4,379.22;实测值380.2(m h)。
[0190]
化合物9:将diea(40μl,0.22mmol)加至缬氨酸-瓜氨酸-pab(oh)*tfa盐2(100mg,0.2mmol)和fmoc-peg8-nhs酯(167mg,0.22mmol)的无水dmf(2ml)溶液中,搅拌45分钟。反应通过lc/ms进行监测,并于100g c18 aq色谱柱上使用梯度洗脱5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物8(145mg,71%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
52h76
n6o
15
,1024.54;实测值1025.5(m h)。
[0191]
化合物10:在室温下,将dcc(18.6mg,0.09mmol)的二氯甲烷(3ml)室温溶液滴加至fmoc-peg8-缬氨酸-瓜氨酸-pab(oh)9(61.8mg,0.06mmol)、vx-787(24mg,0.06mmol)和dmap(7.2mg,0.06mmol)的无水二氯甲烷(12ml)悬浮液中,混合物搅拌16h。真空除去挥发物,残余物于50g c18 aq色谱柱上使用梯度洗脱5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物10,为蓬松的灰白色固体(50mg,51%收率)。ms(esi,pos.):计算值c
72h93
f2n
11o16
,1405.68;实测值1406.6(m h)。
[0192]
化合物11:将5%哌啶的dmf(0.8ml)溶液加至化合物10(50mg,0.035mmol)的无水dmf(1.6ml)溶液中并搅拌40分钟,然后反应物于50g c18 aq色谱柱上使用梯度洗脱5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物11(43mg,95%收率),为蓬松的灰白色固体。ms(esi,pos.):计算值c
57h83
f2n
11o14
,1183.61;实测值1184.6(m h)。1h-nmr(500mhz;dmso-d6):δ9.95(s,1h),8.48(dd,j=9.6,2.1hz,1h),8.27(s,1h),8.19(s,1h),8.15(d,j=3.6hz,1h),8.10(d,j=7.9hz,1h),7.86(d,j=8.3hz,1h),7.60(dd,j=6.7,0.5hz,1h),7.52(d,j=8.3hz,2h),7.22(d,j=8.4hz,2h),5.97(t,j=4.8hz,1h),5.40(s,2h),5.08(d,j=12.7hz,1h),4.99(d,j=12.5hz,1h),4.75(t,j=6.1hz,1h),4.36(q,j=6.7hz,1h),4.22(t,j=7.9hz,1h),δ3.59(q,j=5.6hz,2h),3.5(s,30h),3.35(t,j=5.7hz,3h),3.01-3.00(m,1h),2.96-2.92(m,2h),2.64(dt,j=9.4,4.4hz,2h),2.39-2.35(m,1h),1.97-1.94(m,2h),1.88(s,1h),1.81-1.57(m,6h),1.50-1.35(m,6h),0.84(dd,j=15.4,6.7hz,6h)。
[0193]
由(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮13合成化合物15,如下文方案3中所述。方案3
[0194]
化合物12:在室温下,将亚硫酰氯(11.8mg,0.1mmol)加至搅拌的fmoc-cap-缬氨酸-瓜氨酸-pab(oh)4(61.2mg,0.086mmol)的无水二氯甲烷(1ml)溶液中,并搅拌1小时。在起始物料耗尽后,真空除去挥发物,将残余物于30g c18 aq色谱柱上使用梯度洗脱5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物12,为蓬松的灰白色固体(38mg,60%收率)。ms(esi,pos.):计算值c
39h49
cln6o6,732.3;实测值733.3(m h)。
[0195]
化合物14:将k2co3(28mg,0.20mmol)和nai(6mg,0.041mmol)加至(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮13(20mg,0.041mmol)和fmoc-cap-缬氨酸-瓜氨酸-pab-cl(40mg,0.052mmol)的dmf溶液(1ml)中,将混合物加热至65℃保持40分钟。将反应冷却至室温并于30g c18 aq色谱柱上使用5-60%mecn/h2o(两者均含0.05%乙酸)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物14,为蓬松的灰白色固体(40mg,83%收率)。ms(esi,pos.):计算值c
63h67
f2n9o
10
s,1179.5;实测值1180.3(m h)。
[0196]
化合物15:将1m tbaf的thf溶液(21μl,0.021mmol)滴加至化合物14(25mg,0.021mmol)的thf(1.5ml)溶液中,混合物于室温下搅拌1.5小时。真空除去挥发物,残余物于30g c18 aq色谱柱上使用5-60%mecn/h2o(两者均含0.05%乙酸)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物15(14mg,70%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
48h57
f2n9o8s,957.4;实测值958.3(m h)。1h-nmr(500mhz;dmsod6):δ10.04(s,1h),8.14(d,j=7.5hz,1h),7.85(d,j=8.0hz,1h),7.61(d,j=8.5hz,2h),7.48(d,j=8.5hz,2h),
7.41(d,j=6.6hz,2h),7.20(d,j=7.7hz,1h),7.14-7.12(t,j=7.5hz,1h),7.07(d,j=7.4hz,1h),6.86(d,j=7.7hz,1h),6.75(t,j=7.0hz,1h),6.02(t,j=4.5hz,1h),5.71(s,1h),5.67(d,j=7.7hz,1h),5.46-5.40(m,3h),5.23(d,j=10.7hz,1h),5.05(d,j=10.7hz,1h),4.47-4.37(m,3h),4.20-4.17(m,1h),4.05(d,j=14.4hz,1h),3.96-3.94(m,1h),3.66-3.63(m,1h),3.21-3.15(m,2h),3.03-2.88(m,4h),2.20-2.09(m,2h),2.00-1.96(m,1h),1.72(s,3h),1.62-1.54(m,3h),1.51-1.41(m,3h),1.38-1.27(m,5h),1.26-1.21(m,3h),0.94(t,j=7.4hz,2h),0.84(dd,j=14.1,6.7hz,6h)。
[0197]
由(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮(13)合成化合物18,如下文方案4所示。方案4
[0198]
化合物17:将亚硫酰氯(9μl,0.122mmol)加至mal-cap-val-cit-pab-oh(16)(35mg,0.061mmol)的无水dcm(3ml)悬浮液中,反应在室温下搅拌1小时。将反应液真空浓缩并与甲苯(4ml)共沸干燥,然后其无需进一步纯化即可用于下一步骤。ms(esi,pos.):计算值c
26h39
cln6o6,590.3;实测值591.3m h)。
[0199]
化合物18:将k2co3(41.5mg,0.3mmol)和碘化钠(9mg,0.06mmol)加至(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮(13)(23mg,0.048mmol)和mal-cap-val-cit-pab-cl(17)(35mg,0.06mmol)的dmf(2ml)溶液中。将反应加热至60℃保持1小时,然后冷却至室温,于30g c18 aq色谱柱上使用5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冻干,得到化合物18(28mg,57%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
52h57
f2n9o
10
s,1037.4;实测值1038.3(m h)。1h-nmr(300mhz;dmso-d6):δ10.00(s,1h),8.08(d,j=7.7hz,1h),7.80(d,j=8.7hz,1h),7.61(d,j=8.6hz,2h),7.49(d,j=8.6hz,2h),7.43-7.39(m,2h),7.21(d,j=7.8hz,1h),7.13-7.06(m,2h),7.01(s,2h),6.87(dd,j=7.8,1.0hz,1h),6.76(td,j=7.2,1.5hz,1h),5.97(t,j=5.6hz,1h),5.72(s,1h),5.68(d,j=7.7hz,1h),5.45-5.42(m,3h),5.24(d,j=10.8hz,1h),5.06(d,j=10.8hz,1h),4.49-4.39(m,3h),4.20(dd,j=8.5,6.8hz,1h),4.06(d,j=14.3hz,1h),
3.96(dd,j=10.7,2.5hz,1h),3.66(dd,j=11.3,2.6hz,1h),3.04-2.89(m,3h),2.15(dt,j=10.1,7.3hz,2h),1.97(q,j=6.8hz,1h),1.70-1.58(m,2h),1.53-1.44(m,5h),1.22(m,3h),0.87-0.81(m,6h)。
[0200]
由(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮(13)合成化合物23,如下文方案5所示。方案5
[0201]
化合物20:将diea(41μl,0.236mmol)加至(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮(13)(30mg,0.044mmol)和化合物19(28.5mg,0.059mmol)的氯仿(0.5ml)溶液中。将反应在室温下搅拌0.5小时,然后在4g硅胶gold色谱柱上使用0-5%meoh/dcm进行纯化,得到化合物20(31mg,75%)为无色固体。ms(esi,pos.):计算值c
30h29
f2n7o7s2,701.2;实测值702.1(m h)。
[0202]
化合物21:将三苯基膦(28.8mg,0.11mmol)加至化合物20(31mg,0.044mmol)的10:1thf/h2o(0.88ml)溶液中。在室温下搅拌24小时后,将反应浓缩至干,残余物于5.5g c18 aq色谱柱上使用5-95%mecn/h2o(两者均含0.05%acoh)进行纯化,得到化合物21(20mg,68%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
30h31
f2n5o7s2,675.2;实测值676.1(m h)。化合物23:将hatu(28.5mg,0.075mmol)和diea(13μl,0.075mmol)加至化合物21(20mg,0.030mmol)和mal-cap-val-oh(22)(27.5mg,0.089mmol)的dmf(1.0ml)溶液中。搅拌1小时后,反应物于30g c18 aq色谱柱上使用5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冻干,得到化合物23(4.5mg,16%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
45h51
f2n7o
11
s2,967.3;实测值968.2(m h)。1h-nmr(500mhz;ch3coch
3-d6):δ7.63(d,j=5.0hz,1h),7.53-7.51(m,1h),7.40(d,j=7.8hz,1h),7.32(dd,j=8.9,8.5hz,1h),7.19(t,j=7.3hz,1h),7.14(t,j=7.6hz,2h),7.07(d,j=4.6hz,1h),6.94(t,j=7.1hz,1h),6.86(s,2h),5.88(s,1h),5.79(d,j=7.7hz,1h),5.67-5.64(m,1h),5.58(d,j=10.2hz,1h),5.36(d,j=10.3hz,1h),4.70(dd,j=9.5,1.8hz,1h),4.63-4.61(m,1h),4.52(dd,j=14.5,5.1hz,2h),4.42-4.41(m,1h),4.17-4.12(m,2h),4.08-4.06(m,2h),3.71(t,j=10.2hz,2h),3.61-3.59(m,1h),3.54-3.42(m,5h),3.05(s,3h),3.02-3.01(m,
1h),2.29(q,j=6.7hz,3h),2.19(m,j=6.3hz,1h),1.67-1.57(m,4h),1.31(m,5h),0.97(dd,j=14.5,6.7hz,6h)。方案6
[0203]
化合物24:在0℃下,将三乙胺(17μl,0.12mmol)、三氟甲磺酸酐(20μl,0.12mmol)和dmap(0.7mg,0.006mmol)加至(r)-12-((s)-7,8-二氟-6,11-二氢二苯并[b,e]硫杂环庚三烯-11-基)-7-羟基-3,4,12,12a-四氢-1h-[1,4]噁嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪-6,8-二酮(13)(29mg,0.06mmol)的dcm(2ml)溶液中。搅拌1小时后,将反应真空浓缩,残余物于4g硅胶gold色谱柱上使用乙酸乙酯/己烷进行纯化,得到化合物24(24mg,66%),为淡黄色固体。ms(esi,pos.):计算值c
25h18
f5n3o6s2,615.1;实测值616.0(m h)。
[0204]
化合物25:将三氟甲磺酸酯24(24mg,0.04mmol)的无水1,4-二氧六环(1ml)溶液装入微波管,并加入氢氧化铵水溶液(0.4ml)。将反应加热至80℃保持48小时。减压除去溶剂,残余物于15.5g c18 aq色谱柱上使用5-95%mecn/水(两者均含0.05%acoh)进行纯化。合并纯级分,冻干,得到化合物25(18.3mg,75%),为蓬松的淡黄色固体。ms(esi,pos.):计算值c
24h20
f2n4o3s,482.1;实测值483.1(m h)。1h-nmr(500mhz;cdcl3):δ7.82-7.80(m,1h),7.12-7.07(m,3h),7.03-7.00(m,2h),6.82(td,j=7.1,2.1hz,1h),6.70(d,j=7.3hz,1h),6.06-6.03(m,1h),5.57(d,j=7.5hz,1h),5.38-5.35(m,2h),4.68(dd,j=13.5,2.4hz,1h),4.52(dd,j=10.0,3.0hz,1h),4.07(d,j=13.8hz,1h),3.92(dd,j=11.0,3.0hz,1h),3.76(dd,j=11.7,3.2hz,1h),3.55(t,j=10.5hz,1h),3.44(td,j=11.9,2.6hz,1h),2.96-2.90(m,1h)。方案7
[0205]
化合物26:在氩气下,将小瓶装入三氟甲磺酸酯24(24.4mg,0.04mmol)的无水dmf(1ml)溶液,加入licl(5.2mg,0.12mmol)、pd(pph3)4(2.3mg,0.002mmol)和et3sih(19μl,0.12mmol)。将反应加热至65℃保持2小时。反应物于15.5g c18 aq色谱柱上使用5-95%mecn/h2o(两者均含0.05%acoh)进行纯化,得到标题化合物26(4.6mg,25%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
24h19
f2n3o3s,467.1;实测值468.1(m h)。1h-nmr(500mhz;cdcl3):δ7.30(d,j=3.1hz,1h),7.18-7.10(m,3h),7.06(d,j=7.8hz,1h),7.02(dd,j=7.6,4.5hz,1h),6.89(t,j=7.3hz,1h),6.71-6.69(m,1h),5.83(dd,j=7.8,3.0hz,1h),5.34-5.30(m,2h),4.68(dd,j=13.5,2.1hz,1h),4.62(dd,j=10.0,3.0hz,1h),4.09(d,j
=13.9hz,1h),3.98(dd,j=11.0,2.9hz,1h),3.81(dd,j=12.0,3.2hz,1h),3.56(t,j=10.6hz,1h),3.44(td,j=11.9,2.6hz,1h),3.01(ddd,j=13.4,12.0,3.4hz,1h)。方案8
[0206]
化合物27:将diea和0.8m h2s的thf溶液(90μl,0.072mmol)加至三氟甲磺酸酯24(15mg,0.024mmol)的dmf(1ml)溶液中。搅拌1小时后,真空除去挥发物,将残余物于5.5g c18 aq色谱柱上使用5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冻干,得到标题化合物27(7.0mg,58%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
24h19
f2n3o3s2,499.1;实测值500.1(m h)。1h-nmr(500mhz;cdcl3):δ7.17-7.11(m,4h),7.01(d,j=6.9hz,1h),6.88-6.83(m,1h),6.65(dd,j=28.7,7.9hz,1h),6.28(s,1h),5.81(d,j=7.5hz,1h),5.36(d,j=8.1hz,1h),5.29(dd,j=28.4,14.0hz,1h),4.74(dd,j=12.8,6.9hz,1h),4.65-4.60(m,1h),4.11(dd,j=13.9,7.8hz,1h),3.99-3.96(m,1h),3.81(t,j=11.2hz,1h),3.56(td,j=10.7,4.4hz,1h),3.50-3.44(m,1h),3.01(t,j=12.7hz,1h)。方案9
酰胺基-peg8-cocl。在单独的小瓶中,将化合物30(110mg,0.138mmol)溶于无水thf(3ml),加入diea(53μl,0.304mmol),然后加入fmoc-n-酰胺基-peg8-cocl的thf(4ml)溶液。1小时后,减压除去溶剂,残余物在teledyne isco上用100g c18 aq色谱柱使用5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。将含有纯产物的级分合并,冷冻,冻干,得到标题化合物31(86mg,基于回收的起始物料为53%收率),为蓬松的灰白色固体。ms(esi,pos.):计算值c
74h89
f2n7o
23
,1481.6;实测值1482.6(m h)。
[0211]
化合物32:在0℃下,向化合物31(86mg,0.058mmol)的meoh(12ml)溶液中加入0.1m naome的meoh(1.16ml,0.116mmol)溶液,然后将反应在0℃下搅拌1小时。通过添加离子交换树脂淬灭反应。滤出树脂,将滤液浓缩,得到化合物32,其不经纯化即可用于下一步骤。
[0212]
化合物33:向化合物32(0.058mmol)的dmf(3ml)溶液中加入5%哌啶的dmf(1.5ml)溶液,将反应搅拌45分钟,然后注入到teledyne isco 50g c18 aq色谱柱上,用5-95%mecn/h2o(两者均含0.05%acoh作为改性剂)进行洗脱。合并纯级分,冷冻,冻干,得到标题化合物33(32mg,两步收率为49%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
53h73
f2n7o
18
,1133.5;实测值1134.4(m h)。
[0213]
化合物34:在室温下,向化合物33(32mg,0.028mmol)的thf(2ml)和水(1ml)溶液中加入0.025mm lioh水溶液(1.1ml,0.028mmol),然后将反应搅拌2小时。减压除去挥发物,残余物在teledyne isco上用gemini 30x 150mm色谱柱,使用梯度洗脱5-95%mecn/h2o(两者均含0.05%acoh作为改性剂)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物34(26mg,82%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
52h71
f2n7o
18
,1119.5;实测值1120.4(m h)。1h-nmr(500mhz;dmso-d6):δ9.19(s,1h),8.47(dd,j=9.8,2.8hz,1h),8.26(m,1h),8.16-8.13(m,2h),8.07(s,1h),7.60(d,j=6.8hz,1h),7.02(d,j=8.3hz,1h),6.93(dd,j=8.3,1.4hz,1h),5.60(s,1h),5.02(q,j=17.4hz,3h),4.75(t,j=6.9hz,1h),4.60(d,j=6.9hz,1h),3.68(m,2h),3.51-3.46(m,32h),2.98(d,j=6.6hz,1h),2.89(t,j=5.3hz,2h),2.68-2.61(m,2h),2.00(s,1h),1.92(s,1h),1.80-1.72(m,4h),1.63-1.61(m,1h),1.53-1.43(m,5h)。方案10
[0214]
化合物35根据来自bioconjugate chemistry(2016),27(10),2549-2557的文献所示程序制备得到。
[0215]
化合物36:将edc-hcl(67mg,0.350mmol)和dmap(34mg,0.278mmol)加至vx-787(100mg,0.250mmol)和醇37(125mg,0.250mmol)的ch2cl2(16ml)混合物中。在环境温度下搅拌22小时后,将反应用乙酸乙酯(20ml)稀释并用h2o(20ml)洗。水层用三份20ml乙酸乙酯萃取。合并的有机层用0.5n hcl水溶液(20ml)洗,然后用饱和nahco3水溶液(20ml)洗,最后用盐水(20ml)洗,经na2so4干燥,过滤,真空浓缩。残余物通过色谱法在12g硅胶上用45-100%乙酸乙酯的己烷溶液纯化,得到化合物58b(149mg,67%收率,82%纯度),其无需进一步纯化即可用于下一步骤。ms(esi,pos.):计算值c
41h42
f2n6o
14
,880.27;实测值881.30(m h)。
[0216]
化合物37:将化合物36(35mg,0.0397mmol)的thf(1.5ml)溶液通入氩气鼓泡5分钟使其脱氧。加入zn粉(102mg,1.56mmol),然后加入甲酸铵(12mg,0.0.190mmol)。将氩气再通入混合物3分钟,然后将反应在氩气下于60℃饼形块中加热24小时。反应过滤,固体用几份thf洗。滤液真空浓缩,然后残余物通过色谱法在4g硅胶上纯化,用40-100%乙酸乙酯的己烷溶液洗脱,得到化合物37(14mg,42%),为淡黄色固体。ms(esi,pos.):计算值c
41h44
f2n6o
12
,850.30;实测值851.30(m h)。
[0217]
化合物38:将草酰氯(8μl,0.0933mol)和dmf(2μl)加至fmoc-n-酰胺基-peg8-酸(30mg,0.0452mmol)的无水ch2cl2(1ml)溶液中。将所得亮黄色溶液在环境温度下搅拌1小时,然后真空浓缩。向所得残余物中加入三份1ml干燥ch2cl2,每次加入后浓缩。在单独的小瓶中,将ipr2net(16μl,0.0918mmol)加至化合物37(20mg,0.0235mmol)的ch2cl2(200μl)悬浮液中。向所得混合物中滴加酰氯(0.0452mmol)的ch2cl2(400μl)溶液。在环境温度下搅拌1小时后,混合物真空浓缩。将粗产物溶于dmso并加载至5.5g c18 aq色谱柱上,用20-100%mecn/h2o(mecn和h2o两种溶剂均含0.05%hoac)进行洗脱。合并含有纯产物的级分,冻干,得
到化合物38(7.3mg,21%收率),为白色固体。ms(esi,pos.):计算值c
75h91
f2n7o
23
,1495.61;实测值1496.60(m h)。
[0218]
化合物39:将0.1m naome/meoh溶液(13μl,0.0013mmol)加至0℃的化合物38(2.0mg,0.00134mmol)的无水甲醇(400μl)溶液中。在冰浴中搅拌4小时后,反应用0.2m hcl/meoh溶液(6.5μl,0.0013mmol)中和,真空浓缩。将ch2cl2(0.5ml,每份)加入3次,每次加入后浓缩混合物。将所得残余物溶于无水dmf(0.2ml)并用10%哌啶/dmf溶液(0.2ml)处理。30分钟后,将反应加载至5.5g c18 aq色谱柱上,用0-100%mecn/h2o(mecn和h2o两种溶剂均含0.1%tfa)进行洗脱。合并含有清洁产物的级分,冻干,得到化合物39的tfa盐(1.1mg,71%),为白色固体。ms(esi,pos.):计算值c
52h73
f2n7o
17
,1105.50;实测值1106.50(m h)。1h nmr(500mhz;cd3od)δ8.57(dd,j=9.5,2.8hz,1h),8.17(s,2h),8.08(d,j=1.8hz,1h),8.00(d,j=4.0hz,1h),7.01(d,j=8.4hz,1h),6.90-6.88(m,1h),5.04(s,2h),4.92(d,j=1.9hz,2h),4.63(d,j=7.9hz,1h),3.91(d,j=3.2hz,1h),3.83-3.74(m,6h),3.66-3.53(m,30h),2.92(dd,j=6.6,3.7hz,2h),2.83(d,j=7.0hz,1h),2.64(td,j=6.4,3.4hz,2h),2.10(t,j=1.4hz,1h),1.96-1.81(m,4h),1.74-1.64(m,2h),1.55-1.50(m,2h)。方案11
[0219]
化合物41a:一般方法a:向fmoc-phe-osu(40a)(107mg,0.22mmol)和val-cit-paba tfa盐(2)(99mg,0.2mmol)的dmf(1.5ml)溶液中加入n,n-二异丙基乙胺(105μl,0.6mmol)。将反应在室温下搅拌30分钟,然后注入到50g c18 aq色谱柱上,用梯度5-95%mecn:h2o(两者均含0.05%acoh)进行洗脱。合并纯级分,冷冻,冻干,得到标题化合物41a(23mg,14%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
42h48
n6o7,748.4;实测值749.3(m h)。
[0220]
化合物41b:一般方法b:向fmoc-asp(oallyl)-oh(fmoc-asp(o烯丙基)-oh,40b)(83mg,0.21mmol)、val-cit-paba.tfa盐(2)(99mg,0.2mmol)和hoat(41mg,0.3mmol)的dmf(1.5ml)溶液中加入nmm(66μl,0.6mmol)和edci(48mg,0.25mmol)。将反应在室温下搅拌2小时,然后注入到teledyne isco 30g c18 aq色谱柱上,用5-95%mecn/h2o(两者均含0.05%acoh)进行洗脱。合并纯级分,冷冻,冻干,得到标题化合物41b(85mg,56%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
40h48
n6o9,756.3;实测值757.4(m h)。
[0221]
化合物41c:通过一般方法b使用fmoc-his(trt)-oh(40c)(130mg,0.21mmol)制备得到。产量=146mg(74%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
58h60
n8o7,980.5;
实测值981.4(m h)。
[0222]
化合物41d:通过一般方法a使用fmoc-glu(oallyl)-osu(fmoc-glu(o烯丙基)-osu,40d)(51mg,0.1mmol)制备得到。产量=25mg(33%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
27h26
n2o8,506.2;实测值507.2(m h)。
[0223]
化合物41e:向val-cit-paba(380mg,1.0mmol)的无水dma(5ml)的0℃溶液中加入fmocleuoh(40e)(424mg,1.2mmol)、hoat(170mg,1.2mmol)、edc-hcl(240mg,1.2mmol)和n-甲基吗啉(220μl,2.0mmol)。在冰浴中搅拌2.5小时后,将反应用h2o(35ml)稀释。产物通过真空过滤进行收集并用h2o(5ml,每份)洗3次,然后用6份10-20ml的乙酸乙酯洗,直至lcms分析表明剩余《5%fmocleuoh。真空干燥过夜后,将白色固体粉碎成粉末并再次真空干燥,得到化合物41e(616mg,86%)。ms(esi,pos.):计算值c
39h50
n6o7,714.4;实测值715.4(m h)。
[0224]
化合物41f:向val-cit-paba(200mg,0.527mmol)、fmocarg(pbf)oh(40f)(410mg,0.632mmol)和hoat(86mg,0.632mmol)的无水dmf(5ml)的0℃混合物中加入n-甲基吗啉(90μl,0.818mmol)和edc-hcl(120mg,0.626mmol)。将反应在冰浴中搅拌3小时,然后加载至100g c18色谱柱上,用10-60%mecn/h2o(两种溶剂均含0.05%hoac)进行洗脱。合并含有纯产物的级分,冻干,得到化合物41f(345mg,65%),为白色固体。ms(esi,pos.):计算值c
52h67
n9o
10
s,1009.47;实测值1010.40(m h)。
[0225]
化合物42a:一般方法a:将vx-787(11mg,0.026mmol)、dmap(3.2mg,0.026mmol)和dcc(8mg,0.039mmol)加至fmoc-phe-val-cit-pab-oh(41a)(20mg,0.026mmol)的dcm(3ml)溶液中。搅拌5小时后,加入额外的vx-787(4mg),将反应搅拌16小时,然后真空浓缩。将残余物溶于dmso(1ml)并用teledyne isco在15.5g c18 aq色谱柱上采用梯度洗脱5-95%mecn/h2o(两者均含0.05%acoh)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物42a(10mg,基于回收的起始物料为50%收率),为蓬松的灰白色固体。ms(esi,pos.):计算值c
62h65
f2n
11
o8,1129.5;实测值1130.4(m h)。亦回收了未反应的41a(7mg)。
[0226]
化合物42b:一般方法b:将edci(13.2mg,0.069mmol)加至化合物41b(35mg,0.046mmol)、vx-787(18.5mg,0.046mmol)和dmap(5.6mg,0.046mmol)的dcm(6ml)溶液中,将反应在室温下搅拌2h。加入额外的vx-787(6mg),将反应搅拌过夜。减压除去溶剂,将残余物溶于dmf(1.5ml)并于teledyne isco上用30g c18 aq色谱柱采用5-95%mecn/h2o(两者均含0.05%tfa)进行纯化。合并纯级分,冻干,得到标题化合物42b(23mg,44%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
60h65
f2n
11o10
,1137.5;实测值1138.4(m h)。
[0227]
化合物42b’:将pd(pph3)4(2.4mg,0.002mmol)和苯基硅烷(1滴)加至化合物42b(23mg,0.020mmol)的thf(0.8ml):dmf(0.4ml)溶液中,将反应搅拌30分钟。真空除去挥发物,得到化合物42b’,其不经纯化即可用于下一步骤。
[0228]
化合物42c:使用化合物41c(39mg,0.04mmol),按照一般方法b合成,得到标题化合物42c(30mg,55%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
78h77
f2n
13
o8,1361.6;实测值1362.4(m h)。
[0229]
化合物42d:使用化合物41d(15.4mg,0.02mmol),按照一般方法a合成,得到标题化合物42d(13mg,57%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
61h67
f2n
11o10
,1151.5;实测值1152.4(m h)。
[0230]
化合物42d’:将pd(pph3)4(1.3mg,0.001mmol)和苯基硅烷(2μl,0.0165mmol)加至
化合物42d(13mg,0.011mmol)的thf(1.5ml)/dmf(0.5ml)溶液中,将反应搅拌20分钟。真空除去挥发物,得到化合物42d’,其不经纯化即可用于下一步骤。
[0231]
化合物42e:将fmoc-leu-val-cit-paba(41e)(89mg,0.125mmol)、dmap(30mg,0.245mmol)和edc-hcl(48mg,0.250mmol)加至vx-787(100mg,0.250mmol)的3:1thf:dma(3ml)溶液中。将反应在环境温度下搅拌2小时,然后真空浓缩以除去thf。将剩余的dma溶液加载至50g c18aq色谱柱上,用5-100%mecn/h2o(两种溶剂均含0.05%hoac)进行洗脱。将含有产物的级分冻干,然后通过色谱法在30g c18 aq色谱柱上用5-100%mecn/h2o(两种溶剂均含0.05%hoac)洗脱,进行再纯化。冻干后,得到标题化合物42e(37mg,24%),为白色固体,lcms纯度为~90%。产物不经进一步纯化即可用于下一步骤。ms(esi,pos.):计算值c
59h67
f2n
11
o8,1095.51;实测值1096.50(m h)。
[0232]
化合物42f:将dmap(8mg,0.0655mmol)和edc-hcl(12mg,0.0626mmol)加至化合物41f(31mg,0.0307mmol)和vx-787(25mg,0.0626mmol)的3:1thf:dma(760μl)溶液中。在环境温度下搅拌4小时后,通过真空浓缩除去thf。将剩余的dma溶液加载至15.5g c18 aq色谱柱上,用0-100%mecn/h2o(两种溶剂均含0.05%hoac)进行洗脱。合并含有纯产物的级分,冻干,得到化合物42f(7mg,16%),为白色固体。ms(esi,pos.):计算值c
72h84
f2n
14o11
s,1390.61;实测值1391.45(m h)。
[0233]
fmoc脱去的一般方法:化合物43a:将5%哌啶的dmf(1ml)溶液加至化合物42a(10mg,0.01mmol)的dmf(1ml)溶液中,反应搅拌45分钟。将反应物注入到teledyne isco 15.5g c18 aq色谱柱上,用5-95%mecn:h2o(两种溶剂均含0.05%hoac)进行洗脱。合并纯级分,冷冻,冻干,得到标题化合物43a(7mg,63%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
47h55
f2n
11
o6,907.4;实测值908.4(m h)。
[0234]
化合物43b:按照fmoc脱去的一般方法。由粗制42b’得到的收率=78%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
42h51
f2n
11
o8,875.4;实测值876.4(m h)。
[0235]
化合物43c:按照fmoc脱去的一般方法。由42c得到的收率=53%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
66h67
f2n
13
o8,1139.5;实测值1140.4.(m h)。
[0236]
化合物43d:按照fmoc脱去的一般方法。由42d’得到的收率=59%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
43h53
f2n
11
o8,889.4;实测值890.4(m h)。
[0237]
化合物43e:将10%哌啶的dmf(400μl)溶液加至化合物42e(20mg,0.0182mmol)的无水dmf(400μl)溶液中。在环境温度下搅拌30分钟后,将反应加载至5.5g c18 aq色谱柱上,用5-25%mecn/h2o(两种溶剂均含0.1%tfa)进行洗脱。合并纯级分,冻干,得到化合物43e的tfa盐(13mg,72%),为白色固体。ms(esi,pos.):计算值c
44h57
f2n
11
o6,873.45;实测值874.40(m h)。
[0238]
化合物43f:将10%哌啶/dmf溶液(100μl)加至化合物42f(6.8mg,0.00489mmol)的dmf(100μl)溶液中。在环境温度下搅拌1小时后,将溶液加载至5.5g c18 aq色谱柱上,用0-100%mecn/h2o(两种溶剂均含0.05%hoac)洗脱。合并含有产物的级分,冻干,得到化合物43f(5.2mg,91%),为白色固体。ms(esi,pos.):计算值c
57h74
f2n
14
o9s,1168.55;实测值1169.40(m h)。
[0239]
安装fmoc-n-酰胺基-peg8的一般方法:化合物44a:向化合物43a(7mg,0.008mmol)的dmf(1ml)溶液中加入fmoc-n-酰胺基-peg8-nhs酯(7mg,0.009mmol),随后加入n,n-二异
丙基乙胺(4μl,0.023mmol),然后将反应搅拌2小时。产物在teledyne isco上用15.5g c18 aq色谱柱采用梯度洗脱5-95%mecn/h2o(两者均含0.05%hoac)进行纯化。合并纯级分,冷冻,冻干,得到标题化合物44a(5mg,42%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
81h102
f2n
12o17
,1552.7;实测值1553.7(m h)。
[0240]
化合物44b:按照安装fmoc-n-酰胺基-peg8的一般方法。由43b得到的收率=61%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
76h98
f2n
12o19
,1520.7;实测值1521.4(m h)。
[0241]
化合物44c:按照安装fmoc-n-酰胺基-peg8的一般方法。由43c得到的收率=83%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
97h114
f2n
14o17
,1784.4;实测值1785.6(m h)。
[0242]
化合物44c’:向化合物44c(17mg,0.0073mmol)的dcm(2ml)溶液中加入tfa(0.2ml),搅拌3小时后,反应用meoh(3ml)稀释,真空除去挥发物。将残余物溶于1:1mecn:h2o(1.5ml)和dmso(0.2ml)并注入到teledyne isco 15.5g c18 aq色谱柱上,使用梯度洗脱5-95%mecn:h2o(两者均含10mmol醋酸铵作为改性剂)进行洗脱。合并纯级分,冷冻,冻干,得到标题化合物44c’(12mg,82%),为蓬松的灰白色固体。ms(esi,pos.):计算值c
78h100
f2n
14o17
,1542.7;实测值1543.8(m h)。
[0243]
化合物44d:通过以下一般方法使用43d制备得到。产物44d不经纯化即可用于下一步骤。
[0244]
化合物44e:将ipr2net(15μl,0.0861mmol)加至化合物43e-tfa盐(20mg,0.0206mmol)和fmoc-n-酰胺基-peg8-nhs酯(20mg,0.0263mmol)的无水dma(400μl)溶液中。在环境温度下搅拌2小时后,将反应加载至15.5g c18 aq色谱柱上,用10-100%mecn/h2o(两种溶剂均含0.1%tfa)进行洗脱。合并纯级分,冻干,得到化合物44e(13mg,42%),为白色固体。ms(esi,pos.):计算值c
78h104
f2n
12o17
,1518.76;实测值1519.70(m h)。
[0245]
化合物44f:向化合物43f(7mg,0.00599mmol)的无水dmf(100μl)溶液中加入10%ipr2net的dmf(33μl)溶液,然后加入fmoc-n-酰胺基-peg8-nhs酯(6mg,0.00789mmol)的无水dmf(100μl)溶液。1小时后,lcms表明反应不完全。加入额外的fmoc-n-酰胺基-peg8-nhs酯(25μl的6%dmf溶液),将反应再搅拌2小时以完成反应。通过色谱法在5.5g c18 aq isco色谱柱上用0-100%mecn/h2o(两种溶剂均含0.05%hoac)进行纯化,然后将纯级分冻干,得到化合物44f(9mg,82%),为白色固体。ms(esi,pos.):计算值c
91h121
f2n
15o20
s,1814.0;实测值1815.7(m h)。
[0246]
化合物45a:按照fmoc脱去的一般方法,由44a制备得到标题化合物45a,收率为93%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
66h92
f2n
12o15
,1330.7;实测值1331.6(m h)。1h-nmr(500mhz;dmso-d6):δ10.02(d,j=0.7hz,1h),8.48(dd,j=9.8,2.8hz,1h),8.26(d,j=1.3hz,1h),8.18(s,1h),8.14(d,j=3.8hz,1h),8.08(s,1h),7.85(s,1h)7.60(d,j=6.8hz,1h),7.52(d,j=8.4hz,2h),7.23-7.18(m,6h),7.18-7.14(m,1h),5.99(s,1h),5.42(s,2h),5.07(d,j=12.4hz,1h),4.98(d,j=12.4hz,1h),4.74(t,j=6.5hz,1h),4.60-4.56(m,1h),4.38-4.35(m,1h),4.20(t,j=7.5hz,1h),3.48-3.47(m,25h),3.41(m,2h),3.39-3.34(m,4h),3.02-2.96(m,6h),2.77-2.74(m,1h),2.65-2.62(m,3h),2.37-2.33(m,1h),2.28(t,j=6.1hz,2h),1.97-1.94(m,3h),1.80-1.60(m,6h),1.50-1.36(m,6h),0.84(dd,j=15.0,6.7hz,6h)。
[0247]
化合物45b:按照fmoc脱去的一般方法,由44b制备得到标题化合物45b,收率为
69%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
61h88
f2n
12o17
,1298.6;实测值1299.5(m h)。1h-nmr(500mhz;dmso-d6):δ9.33(s,1h),9.16(dd,j=6.3,0.8hz,1h),8.48(dd,j=9.8,2.8hz,1h),8.42-8.41(m,1h),8.26(d,j=1.3hz,1h),8.18(s,1h),8.14(d,j=3.9hz,1h),7.92(d,j=7.6hz,1h),7.71-7.70(m,2h),7.61-7.60(m,1h),7.30(s,1h),7.21(d,j=8.5hz,2h),5.03(m,2h),4.74-4.70(m,2h),4.02(d,j=1.0hz,1h),3.93-3.91(m,1h),3.60-3.53(m,3h),3.53-3.42(m,35h),2.99-2.95(m,3h),2.81-2.79(m,2h),2.37-2.34(m,2h),2.26-2.23(m,2h),1.97-1.92(m,3h),1.80-1.71(m,3h),1.65-1.61(m,2h),1.52-1.42(m,6h),1.22(d,j=0.3hz,1h),0.92(dd,j=9.6,7.1hz,6h)。
[0248]
化合物45c:按照fmoc脱去的一般方法,由44c’制备得到标题化合物45c,收率为89%,为蓬松的灰白色固体。ms(esi,pos.):计算值c
63h90
f2n
14o15
,1320.7;实测值1322.0(m h)。1h-nmr(500mhz;dmso-d6):δ12.35(d,j=0.9hz,1h),10.03(s,1h),8.95(s,1h),8.47(dd,j=9.7,2.8hz,1h),8.29(d,j=7.6hz,2h),8.24(dd,j=5.3,2.3hz,2h),8.20(d,j=4.0hz,1h),7.83(d,j=8.3hz,2h),7.75-7.69(m,2h),7.51(d,j=8.5hz,2h),7.32(s,1h),7.22(d,j=8.6hz,2h),6.09-6.07(m,1h),5.03(m,2h),4.78-4.75(m,1h),4.69(q,j=7.1hz,1h),4.40-4.37(m,1h),4.22-4.19(m,2h),3.50(s,16h),3.50(s,5h),3.48(d,j=4.2hz,7h),3.46-3.41(m,5h),3.07-2.90(m,8h),2.37-2.34(m,2h),2.00-1.94(m,3h),1.80-1.70(m,3h),1.70-1.56(m,3h),1.51-1.35(m,6h),0.84(dd,j=17.7,6.8hz,6h)。
[0249]
化合物45d:按照fmoc脱去的一般方法,由未经纯化的44制备得到标题化合物45d,收率为47%(经过两步),为蓬松的灰白色固体。ms(esi,pos.):计算值c
62h90
f2n
12o17
,1312.7;实测值1313.6(m h)。1h-nmr(500mhz;dmso-d6):δ10.01(s,1h),8.56(s,1h),8.49-8.47(m,1h),8.30-8.26(m,2h),8.18-8.14(m,2h),7.90(d,j=8.7hz,1h),7.60(d,j=6.7hz,1h),7.53(d,j=8.0hz,2h),7.21(d,j=8.0hz,2h),6.65(s,1h),5.68(s,2h),5.02(m,2h),4.74(t,j=5.7hz,1h),4.28(t,j=6.8hz,2h),4.18(dd,j=8.1,5.6hz,1h),3.51(m,32h),2.95(d,j=5.7hz,4h),2.75-2.74(m,2h),2.42-2.29(m,3h),2.11-2.04(m,3h),1.97-1.93(m,2h),1.82-1.61(m,8h),1.49-1.37(m,6h),1.22(s,3h),0.82(dd,j=12.6,6.6hz,6h)。
[0250]
化合物45e:将10%哌啶的dmf(100μl)溶液加至化合物44e(16mg,0.0153mmol)的dmf(100μl)溶液中。在环境温度下搅拌30分钟后,将反应加载至5.5g c18 aq色谱柱上,用5-100%mecn/h2o(两种溶剂均含0.1%tfa)进行洗脱。将干净的级分冻干,得到化合物45e的tfa盐(7mg,47%),为白色固体。ms(esi,pos.):计算值c
63h94
f2n
12o15
,1296.69;实测值1297.60(m h)。1h nmr(500mhz;dmso-d6)δ10.00(br s,1h),8.48(dd,j=9.8,2.6hz,1h),8.27(m,1h),8.20(s,1h),8.16-8.14(m,1h),8.11-8.04(m,2h),7.71-7.69(m,1h),7.63-7.61(m,1h),7.52(d,j=8.2hz,2h),7.22(d,j=8.9hz,2h),6.55(br s,1h),5.98-5.96(m,1h),5.42(s,2h),5.07(d,j=12.3hz,1h),4.98(d,1h),4.75-4.72(d,j=12.5hz,1h),4.37-4.32(m,2h),4.16(t,j=7.7hz,1h),3.58-3.43(m,32h),3.04-2.93(m,4h),2.69(t,j=5.6hz,2h),2.43-2.29(m,2h),1.97-1.92(m,3h),1.83-1.31(m,16h),0.86-0.78(m,12h)。
[0251]
化合物45f:向化合物44f(4mg,0.0022mmol)的ch2cl2(50μl)悬浮液中加入三异丙基硅烷(5μl),然后加入tfa(50μl)。在环境温度下搅拌90分钟后,反应物真空浓缩。残余物
用ch2cl2(200μl)稀释并浓缩。用ch2cl2(200μl,每份)将产品再稀释3倍,每次均进行浓缩。用5%哌啶的dmf(100μl)溶液处理所得黄色泡沫(化合物44f’)。在环境温度下搅拌1小时后,将反应加载至5.5g c18 aq色谱柱上,用0-100%mecn/h2o(两种溶剂均含0.1%tfa)进行洗脱。合并含有纯产物的级分,冻干,得到化合物45f的tfa盐(1mg,47%),为白色固体。ms(esi,pos.):计算值c
63h95
f2n
15o15
,1339.7;实测值1340.5(m h)。1h nmr(500mhz;dmso-d6)δ12.25(br s,1h),10.00(br s,1h),8.48(dd,j=9.7,2.8hz,1h),8.27(dd,j=2.6,1.3hz,1h),8.19(d,j=2.7hz,1h),8.14(d,j=3.4hz,1h),8.13(br d,j=7hz,1h),8.09(br d,j=7hz,1h),7.76(br s,2h),7.74(br d,j=9hz,1h),7.61(br d j=7hz,1h),7.55-7.50(m,2h),7.22(d,j=8.5hz,2h),6.05(br s,1h),5.47(br s,2h),5.08(d,j=12.5hz,1h),4.99(d,j=12.3hz,1h),4.76-4.73(m,1h),4.38-4.33(m,2h),4.21-4.18(m,1h),3.59-3.54(m,12h),3.49(t,j=10.5hz,30h),3.10-3.03(m,3h),2.96-2.95(m,6h),2.41-2.34(m,2h),2.00-1.94(m,3h),1.80-1.62(m,2h),1.53-1.28(m,6h),0.85-0.81(m,6h)。实施例2:抗血凝素非细胞毒性抗体-药物偶联物合成
[0252]
如下所述合成抗血凝素非细胞毒性抗体-药物偶联物。
[0253]
将抗血凝素(抗ha)单克隆抗体mab11729进行突变以在重链和轻链的c端引入共有llqga五肽序列。所述突变使得抗体酶促偶联至重链上的最大载量为2(每条重链上为1)。使用在重链c端含有相同共有序列的非ha结合mab(衍生自与传染病无关的免疫抗原)作为非结合同型对照。
[0254]
使用400u/mg mab的pngasef(neb p0704l)在pbs ph 7.4中于37℃下将原生mab11729和同型对照抗体去糖基化过夜。随后,使用自旋过滤器(amicon,30kda截止)将反应混合物缓冲交换至pbs ph 7.4。此方法使得抗体酶促偶联至重链上q295位点处最大载量为2。
[0255]
将第二抗血凝素(n3h2)单克隆抗体mab5385(如j virology 2014,vol 88,7130-7144中所述)进行突变以在重链的c端引入共有elqgp五肽序列或在轻链的c端引入ggggsgelqrp。突变使得抗体酶促偶联至最大载量为2。使用在重链c端或轻链c端含有相同共有序列的非ha结合mab(衍生自与传染病无关的免疫抗原)作为非结合同型对照。
[0256]
在重链c端或q295位点处具有偶联位点的抗体以1mg/ml在pbs ph 7.4中进行偶联。化合物6或11(均包含vx-787,但具有不同的连接体)以超过抗体10-40倍摩尔过量进行添加,酶促反应通过每mg抗体添加14个单位的细菌转谷氨酰胺酶(zedira,t1001)开始,并于37℃下孵育16小时。偶联物通过蛋白a色谱法(pierce蛋白a色谱柱,thermoscientific,产品编号20356)进行纯化。偶联物使用waters acquity uplc通过esi-ms进行分析以确定有效负载:抗体比值(dar)。在c4色谱柱(2.1x 50mm acquity uplc beh蛋白c4,1.7um,300a)上以10分钟梯度(分钟:流动相b的百分比;0:10%、1:10%、5:90%、7:90%、7.2:10%、10:10%),进行色谱分离。流动相a为0.1%甲酸水溶液,流动相b为0.1%甲酸/乙腈溶液。流速设置为0.3ml/min。检测器tof扫描设置为m/z 500-4500,主要参数如下所示(毛细管电压3.0kv;取样锥80v;源偏移100v;源温度150℃;去溶剂化温度450℃;锥气体0l/hr;去溶剂化气体800l/hr)。使用masslynx软件中的maxent函数对光谱进行解卷积。所得分子离子,当根据强度加权时,对应于表3和表4中列出的载量。实际的质谱图如图1和图2所示。或者,为了确定抗体上有效负载的载量,偶联物使用tsk-npr butyl hic(疏水相互作用色谱)
色谱柱在agilent 1260上运行,使用1m磷酸钾ph 8.5与水的线性梯度持续60分钟。有效负载载量由对应于经偶联和未偶联抗体种类的峰面积积分来确定。有效负载:抗体比值报告在表5中。体积排除hplc确定所有偶联物均》92%单体(表5)。此方法生成了mab11729-vx-787非细胞毒性抗体-药物偶联物(ncadc),其中mab11729经由所述抗体重链c端的llqga五肽偶联至化合物11(“11729-hc-c端-11”);mab11729-vx-787ncadc,其中mab11729经由所述抗体重链n端的llqga五肽偶联至化合物11(“11729-hc-n端-11”);mab11729-vx-787ncadc,其中mab11729经由所述抗体轻链c端的llqga五肽偶联至化合物11(“11729-lc-c端-11”);mab11729-vx-787ncadc,其中mab11729经由所述抗体轻链n端的llqga五肽偶联至化合物11(“11729-lc-n端-11”);mab11729-vx-787ncadc,其中化合物11在q295位点处进行偶联(“11729-q295-11”);同型对照抗体,其中化合物11经由llqga五肽在所述重链c端进行偶联(“同型对照-hc-c端-11”);同型对照抗体,其中化合物11在q295位点处进行偶联(“同型对照-q295-11”);mab11729-vx-787ncadc,其中化合物6在q295位点处进行偶联(“11729-q295-6”);同型对照抗体,其中化合物6经由llqga五肽在所述重链c端进行偶联(“同型对照-hc-c端-6”);同型对照抗体,其中化合物6在q295位点处进行偶联(“同型对照-q295-6”);mab11729-巴洛沙韦ncadc,其中化合物15经由llqga五肽在所述mab11729重链c端进行偶联(“11729-hc-c端-15”);和同型对照抗体,其中化合物15在所述重链c端进行偶联(“同型对照-hc-c端-15”)。
[0257]
原生mab11729和同型对照抗体(1-10mg/ml)于50mm hepes、150mm nacl、ph 7.5中在37℃下,用1mm二硫苏糖醇处理30分钟。凝胶过滤(g-25,ph 4.5醋酸钠)后,将马来酰亚胺基连接体-有效负载衍生化合物18(1.2当量/sh基团)的dmso溶液(10mg/ml)加至经还原的抗体中,并将混合物用1m hepes(ph 7.4)调节至ph 7.0。偶联物使用含5%甘油的pbs通过体积排除色谱法进行纯化,然后无菌过滤。蛋白质浓度以及有效负载与抗体比值均通过uv光谱分析确定。体积排除hplc确定所使用的所有偶联物均》95%单体。所有经偶联的抗体的连接体-有效负载载量值均通过质谱进行分析。有效负载与抗体比值报告于表6中。
[0258]
表3、表4和表5包含某些偶联物的载量数据(esi-ms dar)。经转谷氨酰胺酶偶联的化合物11和15产生的dar接近理论值2。表6包含ncadc的纯度及dar值列表。表3. 11729-q295-11和同型对照-q295-11中强度加权平均有效负载载量汇总11中强度加权平均有效负载载量汇总表4. 11729-hc-c端-11和同型对照-hc-c端-11中强度加权平均有效负载载量汇总
表5. 11729-hc-c端-15和同型对照-hc-c端-15中强度加权平均有效负载载量汇总表6.化合物6、11和15偶联物的纯度(经由sec)和dar
hc-c端-11。
[0260]
将抗体在startingblock封闭缓冲液中稀释至100μg/ml的起始浓度,每种抗体以1:4滴定至最终浓度为6.1x10-3
μg/ml。将板孵育后,去除startingblock封闭缓冲液,将经稀释的抗体以75μl/孔添加至细胞。将板在室温下孵育1小时。孵育后,将板用清洗缓冲液(咪唑缓冲盐水和20在milli-q水中稀释至1倍;kpl)洗3次,并用在startingblock封闭缓冲液中以1:2000稀释的75μl/孔的二抗(驴抗人igg hrp-偶联;jackson immunoresearch)覆盖。将此第二溶液在板上于室温下温育1小时。随后,用清洗缓冲液将板洗3次,然后以1:1的比例加入75μl/孔的elisa pico化学发光底物。立即在molecular devices spectramax i3x酶标仪上读取板的发光情况。
[0261]
11729-hc-c端-11、11729-lc-c端-11和11729-lc-n端-11均以亚纳摩尔浓度与甲型流感病毒感染的细胞结合,表现出特异性结合,如表7和图3所示。值得注意的是,此种特异性结合与未偶联的mab11729抗体在同一范围内,表明添加有效负载对结合不存在过度有害的影响。连接体-有效负载与所述重链n端的偶联使得与甲型流感/h1n1/pr8感染细胞的结合减少,但连接体-有效负载与所述重链c端的偶联则仍然保持与甲型流感/h1n1/pr8感染细胞的结合,如图11所示。表7.甲型流感感染细胞抗ha结合elisa抗体ic
50 log[m]未偶联同型对照抗体未检测到结合mab117291.817x10-9
同型对照-hc-c端-11未检测到结合11729-hc-c端-113.756x10-9
[0262]
为了测试抗病毒功效,检测了11729-hc-c端-11抑制流感病毒感染细胞的能力。mdck london细胞以20,000个细胞/孔接种在96孔板中的100μl生长培养基(dmem,含有1%丙酮酸钠、10%胎牛血清和0.5%庆大霉素)中。将细胞于37℃、5%co2下孵育18小时。次日,所有抗体均在胰蛋白酶感染培养基(dmem,含有1%丙酮酸钠、0.21%低igg bsa溶液、1mg/ml胰蛋白酶tpck处理和0.5%庆大霉素)中稀释至500μg/ml的起始浓度,然后以1:3滴定至终浓度为1.143x10-1
μg/ml。h1n1 a/puerto rico/08/1934流感病毒,被设计为在其感染的细胞中表达gfp(“h1n1 a/puerto rico/08/1934-gfp”),在胰蛋白酶感染培养基(life technologies)中稀释至moi为1,并与经稀释的抗体或adc以1:1的比例混合。从接种的96孔板中除去生长培养基,并将病毒-抗体或病毒-adc混合物以每孔100μl添加至细胞。将板轻轻敲击并放回至37℃、5%co2下保持20小时。随后,将板用pbs清洗1次,并用50μl 4%pfa的pbs溶液固定,于室温下孵育15分钟。板用pbs洗两次,并用50μl的pbs覆盖。立即在immunospot分析仪(cellular technology limited)上读取板的gfp信号。
[0263]
如表8和图4所示,连接体-有效负载11与重链和轻链的c端以及与轻链的n端的偶联使得抗体-药物偶联物对抗甲型流感/h1n1/pr8病毒的抗病毒活性相对于亲本抗体提高了3至163倍。如表9和图6所示,连接体-有效负载11与重链c端的偶联使得抗体-药物偶联物对抗甲型流感/h1n1/cal09病毒的抗病毒活性相对于亲本抗体提高了10倍。如表8和图7所示,连接体-有效负载6与重链c端的偶联使得抗体-药物偶联物对抗甲型流感/h1n1/pr8病毒的抗病毒活性相对于亲本抗体提高了51倍;然而,6与轻链c端的偶联相对于亲本抗体并
未增加抗病毒活性。如表8和图8所示,对于11729-hc-c端-34,连接体-有效负载与重链c端的偶联使得抗体-药物偶联物对抗甲型流感/h1n1/pr8病毒的抗病毒活性相对于亲本抗体提高了7倍,而对于1129-hc-c端-45a则提高了35倍,但连接体-有效负载与轻链c端的偶联相对于亲本抗体并未增加抗病毒活性。如表9和图9所示,连接体-有效负载与轻链和重链的c端的偶联使得抗体-药物偶联物对抗甲型流感/h3n2/hk68x31病毒的抗病毒活性相对于亲本抗体提高了3至10倍。如表8和图10所示,连接体-有效负载与重链c端的偶联使得抗体-药物偶联物对抗甲型流感/h1n1/pr8病毒的抗病毒活性相对于亲本抗体提高了71倍。表8.抗甲型流感/h1n1/pr8病毒抗体的抗病毒活性表8.抗甲型流感/h1n1/pr8病毒抗体的抗病毒活性表9.抗甲型流感/h1n1/cal09病毒抗体的抗病毒活性抗体ic
50 log[m]相对于亲本抗体增加倍数mab117297.022x10-10
1.0011729-hc-c端-116.528x10-11
10.76
同型对照-hc-c端-11未检测到功效n/a表10.抗甲型流感/h3n2/hk68x31病毒抗体的抗病毒活性抗体ic
50 log[m]相对于亲本抗体增加倍数mab53855.15x10-10
1.005385-hc-c端-115.262x10-11
9.785385-lc-c端-111.802x10-10
2.86同型对照-hc-c端-11未检测到功效n/a同型对照-lc-c端-11未检测到功效n/a同型对照抗体未检测到功效n/avx-7877.045x10-9
n/a实施例4:抗流感ha mab11729抗体-药物偶联物在猴或igg耗尽的人血浆中的体外血浆稳定性
[0264]
11729-hc-c端-11在体外与来自不同物种的血浆一同温育并对dar进行了评估。
[0265]
将ncadc样品分别独立地添加至新鲜汇集的食蟹猴血浆(bioreclamation,产品批号cyn260056-cyn260057)或igg耗尽的人血浆(bioivt,产品批号brh1097869)中,在eppendorf管(eppendorf,目录号022363514)中最终浓度分别为50μg/ml,然后于37℃水浴中孵育0-72小时。分别在0、24、48和72小时时取出样品,然后于-80℃冷冻保存直至用于分析。
[0266]
对于dar分析,使用dynamag-2磁性粒子处理器(life technologies,目录号12321d)通过亲和捕获从血浆样品纯化ncadc。首先,将生物素化的抗人κ抗体(regeneron生成的试剂)固定在链霉亲和素顺磁珠(invitrogen,目录号605602)上。在thermomixer c装置(eppendorf,目录号2231000574)中,于室温下以1850rpm的速度将含有ncadc的各血浆样品与1mg珠粒混合2小时。然后用600μl的50mm tris-hcl ph 7.5缓冲液(由1m tris-hcl ph 7.5缓冲液稀释,invitrogen,目录号15567-027)清洗珠粒3次,然后用600μl的10%乙腈(vwr chemicals,目录号bdh83640.100e)水溶液清洗一次。清洗后,通过将珠粒与50μl 1%甲酸的25:75乙腈:水(v/v)溶液在室温下一同孵育15分钟来洗脱ncadc。通过向每个样品(19.2mm tcep的最终溶液)添加2μl 0.5m tcep(sigma,目录号646547-10x1ml)来进一步减少洗脱的样品,并在thermomixer c装置中于50℃下孵育30分钟。
[0267]
将经还原的ncadc样品注入至与synapt g2-si质谱仪(waters)耦合的1.7μm beh300 c4色谱柱(waters corporation,目录号186007567)上。流速为8μl/min(流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸/乙腈),液相色谱梯度为10分钟梯度,ncadc在2.1-6.5分钟内洗脱,相当于流动相b的26-40%。
[0268]
使用maxent1软件(waters corporation)对所得光谱进行解卷积,参数如下:质量范围:20-60kda m/z,范围:700da-4000 da;分辨率:1.0da/通道;半高宽度:1.0da;最小强度比:33%;最大迭代次数:15。
[0269]
在与食蟹猴血浆或igg耗尽的人血浆孵育72小时后,未观察到来自ncadc的连接体-有效负载的显著损失(图5)。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。