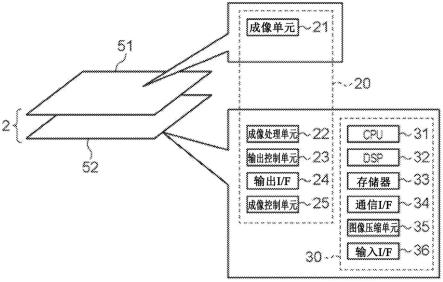
一种腈类化合物的制备工艺
(一)技术领域
1.本发明涉及腈类化合物的制备工艺,特别涉及利用氟磺酸咪唑盐作为促进剂,使醛肟类化合物发生β消除反应一锅法生成腈类化合物的制备工艺。
(二)
背景技术:
2.腈类化合物作为一类较高应用价值的化合物,不仅是合成染料、医药和农用化学品等多用途的中间体,而且还是转化为酰胺、羧酸、胺、酮和酯的关键前体。与使用剧毒氰化物合成腈类化合物这一经典构建氰基的策略相比,经醛肟脱水制得腈类化合物这一方法具有较大的优势,因为醛肟易于获得,反应过程中避免了剧毒氰化物的使用,并产生水作为副产物。虽然已经报道了许多醛肟脱水形成腈的方法,但仍存在一定的局限性。例如,2021年,mary p.watson等报道了以胺为原料,katritzky吡啶鎓盐作促进剂,氰化锌作为氰源,同时加入金属催化剂nicl2和配体xantphos,在添加剂et2zn和znbr2存在条件下,二甲基亚砜溶剂中80℃反应16h,合成了腈类化合物[org.lett.,2021,23,6242
–
6245.];gerald b.hammond课题组于2020年报道了以醛类化合物为原料,盐酸羟胺作为氮源,hcl
·
dmpu作促进剂同时作溶剂,60℃下反应4小时得到目标腈类化合物[green chem.,2020,22,4161-4164.];2019年,秦教授开发了一种在(nh2oh/na2co3/so2f2)体系下直接将醛转化为腈的方法[j.org.chem.,2019,84,5803
–
5812.]。然而,以上方法都存在一些缺点,如需要剧毒氰化物和昂贵的金属催化剂,高温或较高的反应温度以及较长的反应时间,需要添加额外化学计量的化学试剂,此外,温室气体(so2f2)的反应体系对环境不友好,不易操作,不利于大规模应用。
[0003]
氟磺酰咪唑盐(结构如式a所示)作为一种稳定、易存储运输的固体类化合物,由董佳家教授等人首次报道合成,并将其作为高效的磺酰氟试剂,在室温和碱条件下即可完成对苯酚、杂芳基酚的氟磺酰化。该反应表现出了非常好的兼容性和效率,且不需过渡金属参与等优点[angew.chem.,int.ed.,2018,57,2605.]。
[0004][0005]
但是现有技术中未有采用氟磺酰咪唑盐制备腈类化合物的报道,需要寻找一种高效、环保、经济的合成腈类化合物的方法。
(三)
技术实现要素:
[0006]
本发明针对现有技术中存在的缺陷,提供一种高效、环保、经济的合成腈类化合物的制备工艺,本发明采用价廉易得的醛肟作为原料,以稳定、易储存的氟磺酰咪唑盐(a)作为促进剂,经β消除得到腈类化合物,操作简单,反应高效,收率得到了显著提高。
[0007]
本发明采用的技术方案是:
[0008]
本发明提供一种腈类化合物的制备工艺,所述制备工艺包括如下步骤:
[0009]
以式(i)所示醛肟类化合物为原料,加入氟磺酰咪唑盐、溶剂、碱,于20~50℃下反
应0.1~6h,反应液分离纯化,制得(ⅱ)所示的腈类化合物;所述碱为下列之一:碳酸氢钾(khco3)、碳酸氢钠(nahco3)、碳酸钠(na2co3)、碳酸钾(k2co3)、磷酸三钾(k3po4)1,8-二氮杂二环十一碳-7-烯(dbu)、三乙胺(et3n)或二异丙基乙胺(dipea);所述溶剂为下列之一:水、甲醇、乙醇、二氯甲烷、乙腈、乙酸乙酯、二氧六环、甲苯、四氢呋喃、二甲基亚砜或n,n-二甲基甲酰胺;
[0010][0011]
式(i)中r为芳香基、c1-c14的直链或支链烷基,式(ii)中r与式(i)中r相同。
[0012]
进一步,所述式(i)中r为p-meoph(对甲氧基苯基)、p-brph(对溴苯基)、2-萘基、4-coomeph(4-甲酸苯基)或4-联苯基。
[0013]
进一步,所述溶剂体积用量以式(i)所示腈类化合物物质的量计为1~30ml/mmol,优选1-3ml/mmol。
[0014]
进一步,所述氟磺酰咪唑盐与式(i)所示醛肟类化合物的物质的量之比为1~5:1,优选1.5:1。
[0015]
进一步,所述碱与式(i)所示醛肟类化合物的物质的量之比为1~5:1,优选2-3:1。
[0016]
进一步,所述反应温度优选为25~30℃,反应时间优选为10min。
[0017]
进一步,所述反应液分离纯化方法为:反应结束后,加入乙酸乙酯和水萃取,有机相经饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到所述的产物。所述乙酸乙酯和水体积比为1:1,所述乙酸乙酯和水总体积用量以式(i)所示醛肟类化合物物质的量计为1-30ml/mmol,优选20ml/mmol。
[0018]
与现有技术相比,本发明的有益效果主要体现在:
[0019]
1、本发明使用醛肟作为原料,价廉且易制备。
[0020]
2、本发明使用结构稳定、易于储存的固体氟磺酰咪唑盐(a)作为促进剂,高效促进醛肟发生β消除,生成腈,且该试剂经水洗即可除去。
[0021]
3、醛肟、氟磺酰咪唑盐(a)避免了使用氰化物和硫酰氟等危险试剂,因此可以作为制备腈的标准处理条件的绿色替代品。
[0022]
4、底物适用性广,最高能以99%的收率得到相对应的腈类化合物。
[0023]
5、操作过程简单,后处理只需水洗即可得到目标产物,适合大规模制备。
(四)附图说明
[0024]
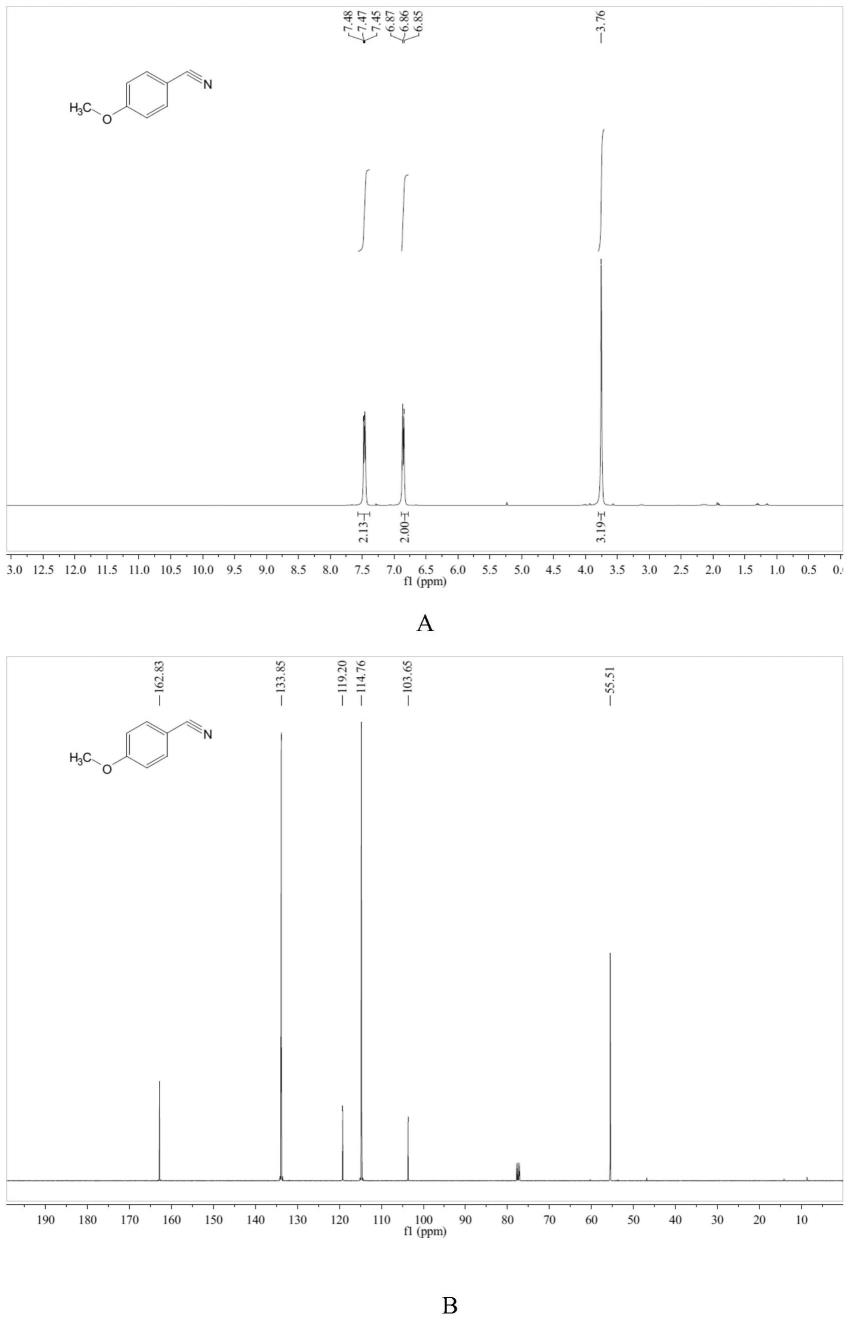
图1为实施例1化合物的核磁共振氢谱(a)、碳谱(b)图。
[0025]
图2为实施例2化合物的核磁共振氢谱(a)、碳谱(b)图。
[0026]
图3为实施例3化合物的核磁共振氢谱(a)、碳谱(b)图。
[0027]
图4为实施例4化合物的核磁共振氢谱(a)、碳谱(b)图。
[0028]
图5为实施例5化合物的核磁共振氢谱(a)、碳谱(b)图。
(五)具体实施方式
[0029]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0030]
实施例1:对甲氧基苯甲腈的制备
[0031]
在一个50ml单口烧瓶中,依次加入对甲氧基苯甲醛肟(式
ⅰ‑
1,r=p-meoph)1.51g(10mmol),20ml乙腈,4.92g(15mmol)氟磺酰咪唑盐(a),2.02g(20mmol)三乙胺,25℃条件下搅拌10min;反应结束后,反应液移入500ml分液漏斗中并加入100ml水和100ml乙酸乙酯萃取,取有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到对甲氧基苯甲腈(式
ⅱ‑
1,r=p-meoph)1.32g,核磁共振氢谱图见图1中a所示,核磁共振碳谱图见图1中b所示,收率99%。
[0032]
核磁共振氢谱:(400mhz,chloroform-d)(δ,ppm):7.57
–
7.38(m,2h),6.88
–
6.77(m,2h),3.76(s,3h).
[0033]
核磁共振碳谱:(101mhz,chloroform-d)(δ,ppm):162.83,133.85,119.20,114.76,103.65,55.51.
[0034][0035]
对照例1:以咪唑盐作促进剂制备对甲氧基苯甲腈
[0036]
在一个50ml单口烧瓶中,依次加入对甲氧基苯甲醛肟(式
ⅰ‑
1,r=p-meoph)1.51g(10mmol),20ml乙腈,3.69g(15mmol)咪唑盐(a’),2.02g(20mmol)三乙胺,25℃条件下搅拌10min;反应结束后,反应液移入500ml分液漏斗中并加入100ml水和100ml乙酸乙酯萃取,取有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,未得到对甲氧基苯甲腈(式
ⅱ‑
1,r=p-meoph)。
[0037][0038]
实施例2:对溴苯甲腈的制备
[0039]
在一个50ml单口烧瓶中,依次加入对溴苯甲醛肟(式
ⅰ‑
2,r=p-brph)2.00g(10mmol),20ml二氯甲烷,4.92g(15mmol)氟磺酰咪唑盐(a),3.03g(30mmol)三乙胺,25℃条件下搅拌10min;反应结束后,反应液移入500ml分液漏斗中并加入100ml水和100ml乙酸乙酯萃取,取有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到对甲氧基苯甲腈(式
ⅱ‑
2,r=p-brph)1.73g,核磁共振氢谱图见图2中a所示,核磁共振碳谱图见图2中b所示,收率95%。
[0040]
核磁共振氢谱:(400mhz,chloroform-d)(δ,ppm):7.62
–
7.54(m,2h),7.52
–
7.45(m,2h).
[0041]
核磁共振碳谱:(101mhz,chloroform-d)(δ,ppm):133.46,132.62,127.97,118.07,111.19.
[0042]
[0043]
实施例3:2-萘甲腈的制备
[0044]
在一个50ml单口烧瓶中,依次加入2-萘甲醛肟(式
ⅰ‑
3,r=2-萘基)1.71g(10mmol),20ml乙酸乙酯,4.92g(15mmol)氟磺酰咪唑盐(a),3.04g(20mmol)dbu,25℃条件下搅拌10min;反应结束后,反应液移入500ml分液漏斗中并加入100ml水和100ml乙酸乙酯萃取,取有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到2-萘甲腈(式
ⅱ‑
3,r=2-萘基)1.47g,核磁共振氢谱图见图3中a所示,核磁共振碳谱图见图3中b所示,收率96%。
[0045]
核磁共振氢谱:(400mhz,chloroform-d)(δ,ppm):8.15(s,1h),7.85(t,j=9.1hz,3h),7.67
–
7.51(m,3h).
[0046]
核磁共振碳谱:(101mhz,chloroform-d)(δ,ppm):134.60,134.11,132.18,129.20,129.09,128.40,128.06,127.69,126.27,119.31,109.27.
[0047][0048]
实施例4:4-氰基苯甲酸甲酯的制备
[0049]
在一个50ml单口烧瓶中,依次加入2-萘甲醛肟(式
ⅰ‑
4,r=4-coomeph)1.79g(10mmol),20ml甲醇,4.92g(15mmol)氟磺酰咪唑盐(a),2.02g(20mmol)三乙胺,30℃条件下搅拌10min;反应结束后,反应液移入500ml分液漏斗中并加入100ml水和100ml乙酸乙酯萃取,取有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到4-氰基苯甲酸甲酯(式
ⅱ‑
4,r=4-coomeph)1.59g,核磁共振氢谱图见图4中a所示,核磁共振碳谱图见图4中b所示,收率99%。
[0050]
核磁共振氢谱:(400mhz,chloroform-d)(δ,ppm):8.07
–
7.96(m,2h),7.71
–
7.57(m,2h),3.86(s,3h).
[0051]
核磁共振碳谱:(101mhz,chloroform-d)(δ,ppm):165.27,133.81,132.18,129.99,117.88,116.23,52.63.
[0052][0053]
实施例5:4-氰基联苯的制备
[0054]
在一个50ml单口烧瓶中,依次加入2-萘甲醛肟(式
ⅰ‑
5,r=4-联苯基)1.97g(10mmol),20ml二甲基亚砜,4.92g(15mmol)氟磺酰咪唑盐(a),5.30g(50mmol)碳酸钠,50℃条件下搅拌10min;反应结束后,反应液移入500ml分液漏斗中并加入100ml水和100ml乙酸乙酯萃取,取有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到4-氰基联苯(式
ⅱ‑
5,r=4-联苯基)1.77g,核磁共振氢谱图见图5中a所示,核磁共振碳谱图见图5中b所示,收率99%。
[0055]
核磁共振氢谱:(400mhz,chloroform-d)(δ,ppm):7.61(q,j=8.5,7.7hz,4h),7.56(d,j=7.2hz,2h),7.51
–
7.37(m,3h).
[0056]
核磁共振碳谱:(101mhz,chloroform-d)(δ,ppm):145.41,138.98,132.61,129.23,128.81,127.66,127.24,119.06,110.85.
[0057]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。