一种
α-咔啉的制备方法及
α-咔啉
技术领域
1.本发明属于咔啉类衍生物的有机合成技术领域,具体涉及一种α-咔啉的制备方法及α-咔啉。
背景技术:
2.咔啉衍生物是一类具有吡啶并吲哚结构的三环杂芳胺类化合物,是具有抗菌、抗病毒、抗肿瘤等重要生物、药理活性的含氮生物碱。根据吡啶氮原子位置不同,分为α-咔啉、β-咔啉、γ-咔啉和δ-咔啉类化合物。其中,α-咔啉类化合物具有优良的抗病毒、抗癌和抗疟原虫活性,同时在抗抑郁、抗精神疾病、激酶抑制剂和治疗心脑血管疾病中也有重要应用。根据文献报道,目前α-咔啉类化合物主要合成路线有以下几种。
3.(一)3-(2-硝基苯基)吡啶经分子内硝基还原成环法制备α-咔啉。例如,2004年jacqueline h.smitrovich等(org.lett.2004,6,533-535)报道了一种α-咔啉的合成方法;以3-(2-硝基苯基)吡啶为原料,在醋酸钯/邻菲罗啉催化体系、约5atm(5个标准大气压)一氧化碳氛围、dmf中140℃反应16小时,经还原成环反应得到α-咔啉,反应式如下。
[0004][0005]
(二)稠合三氮唑经分子内脱氮、胺环化反应制备α-咔啉。例如,2018年buddhadeb chattopadhyay等(j.am.chem.soc.2018,140,8429-8433)报道了一种α-咔啉的合成方法;以8-苯基四唑并[1,5-a]吡啶为原料,在[ir*cpcl2]2/agsbf6催化体系、苯中130℃反应24小时,经分子内脱氮气、转胺环化得到α-咔啉,反应式如下。
[0006][0007]
(三)2,3-二卤代吡啶与芳胺经分子间c-n偶联、分子内芳基化制备α-咔啉。例如,2009年gregory d.cuny等(j.org.chem.2009,74,3152-3155)报道了一种α-咔啉的合成方法;以2,3-二氯吡啶或2,3-二溴吡啶和苯胺为原料,首先在醋酸钯/三苯基膦催化体系、叔丁醇钠作碱、邻二甲苯中120℃反应3小时,经分子间c-n偶联反应得到3-溴/氯-n-苯基吡啶-2-胺中间体,再在醋酸钯/三环己基膦氟硼酸盐催化体系、dbu作碱、邻二甲苯/n,n-二甲基苯胺中145℃反应16小时,经分子内c-h/c-x偶联关环芳基化反应得到α-咔啉,反应式如下。
[0008]
[0009]
但是上述合成路线均存在一定的问题,具体如下:路线(一)使用3-(2-硝基苯基)吡啶为起始原料,该原料不易得,价格昂贵;同时需要5个大气压的一氧化碳氛围下反应,反应条件苛刻,生产成本高、危险系数大;反应副产物较多,产物不易提纯,极大限制了其在工业化生产中的应用;路线(二)使用8-苯基四唑并[1,5-a]吡啶为起始原料,原料成本昂贵;采用[ir*cpcl2]2/agsbf6的催化体系,且无法回收利用,造成生产本过高;路线(三)使用价格相对低廉的苯胺和2,3-二溴/氯吡啶作原料,原料成本低,经济性好,但该路线反应操作复杂、反应条件苛刻,需要变换反应条件,实际操作不易控制;同时采用邻二甲苯/n,n-二甲基苯胺混合溶剂,无法回收溶剂,环境问题突出,增加了反应成本;此外,其反应选择性差、收率较低,造成生产成本偏高。
[0010]
综上所述,现有合成方法存在原料价格昂贵、不易获得,合成条件要求高,反应选择性、原子经济性和总反应收率低,分离提纯难度大,生产成本高,严重制约了其在制备α-咔啉的工业化应用。因此,有必要对现有技术中α-咔啉的生产工艺进行改进,在确保较高的产物收率的基础上,开发一种更为经济、绿色、高效的α-咔啉的新合成路线。
[0011]
公开于该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域一般技术人员所公知的现有技术。
技术实现要素:
[0012]
为了解决现有技术中α-咔啉的制备成本高、合成条件苛刻、制备困难、环境问题突出、不利于工业化量产的技术问题,提供一种α-咔啉的制备方法及α-咔啉。
[0013]
本发明的发明原理如下:
[0014]
本发明采用镍/铜双金属协同催化,其反应机理推测为铜盐/草酰二胺催化体系促进c-n偶联反应进行生成相应的n-(2-氯苯基)吡啶-2-胺中间体,反应中间体中的吡啶和胺上的氮原子与铜盐配位形成环n-(2-氯苯基)吡啶-2-胺铜盐,从而限制其以胺上氮原子为轴的自由旋转使其吡啶环和苯环的夹角减小,阻止另一分子邻二氯苯与中间体的进一步c-n偶联反应,同时大大促进了吡啶β位上亲电取代反应,所形成的中间态产物中吡啶β位的反应活性增加,进而与镍催化剂生成环镍中间体,随后发生还原消除生成目标产物。
[0015]
本发明的技术方案如下:
[0016]
本发明第一方面提供一种α-咔啉的制备方法,步骤包括:以邻二氯苯和2-氨基吡啶为原料,加入催化剂、配体、碱,经c-n偶联、分子内c-h/c-x交叉偶联串联反应,得到所述α-咔啉,反应式如下:
[0017][0018]
在一些实施方式中,上述催化剂为催化剂a和催化剂b的复合催化剂。
[0019]
在一些实施方式中,上述催化剂a为铜盐;
[0020]
和/或,上述催化剂b为氮杂环卡宾镍盐。
[0021]
在一些实施方式中,上述铜盐选自氧化亚铜、碘化亚铜、溴化亚铜、氯化亚铜中的一种或多种;
[0022]
和/或,上述氮杂环卡宾镍盐选自[ni(ipr*)cpcl]、[ni(ipr*
tol
)cpcl]或[ni(ipr
*
ome
)cpcl]中的一种或多种,对应的结构如下:
[0023][0024]
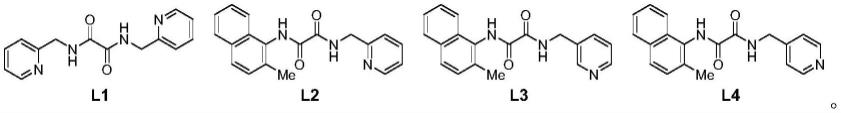
在一些实施方式中,上述配体为草酰二胺配体,其与铜盐构成c-n偶联的常用催化体系,在铜催化邻二氯苯与2-氨基吡啶发生c-n偶联反应过程中,与铜配位形成稳定环铜中间体,促进c-n偶联的进行;优选的,上述草酰二胺配体结构式为如下l1、l2、l3、l4任意一种:
[0025][0026]
在一些实施方式中,上述碱为有机碱,其为c-n偶联反应提供必要的碱性条件,中和反应过程中生成的酸,促进反应正向进行,从而有利于镍催化c-h/c-x分子内偶联反应的进行;优选的,上述有机碱选自叔丁醇金属碱、1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)、胺类化合物中的一种或多种;更优选的,上述有机碱选自叔丁醇锂、叔丁醇钾、叔丁醇钠、1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)、三乙胺中的一种或多种,其中,叔丁醇金属碱能使吡啶β位形成吡啶自由基,更有利于镍催化c-h/c-x分子内偶联反应的进行。
[0027]
在一些实施方式中,上述添加剂选自碱金属卤化物;优选的,上述添加剂选自碱金属卤化物选自溴化钠、溴化钾、溴化锂、碘化钠等中的或多种,其在反应过程中与氯代芳烃发生卤交换,从而得到溴代或碘代芳烃中间态,从而大大加快反应进程、促进反应正向进行,反应更加完全、提高反应选择性和收率。
[0028]
在一些实施方式中,上述溶剂为邻二氯苯。邻二氯苯既为溶剂又为反应原料,避免了杂质的引入,有利于提高收率。
[0029]
在一些实施方式中,上述2-氨基吡啶与上述邻二氯苯的摩尔比为1:(1.5~5.0)。
[0030]
在一些实施方式中,上述2-氨基吡啶、邻二氯苯、催化剂a、催化剂b、配体、碱及添加剂的摩尔比为1:(1.5~5.0):(0.001~0.010):(0.0001~0.005):(0.001~0.010):(2.0~3.0):(0.1~1.0)。
[0031]
在一些实施方式中,上述的制备方法进一步包括对串联反应制得的α-咔啉进行后处理;优选的,上述后处理包括萃取、脱溶和结晶;上述串联反应在80~130℃恒温搅拌条件下反应16~48h。
[0032]
本发明方面提供一种α-咔啉,通过上述制备方法制备得到。
[0033]
相比于现有技术,本发明达到的技术效果如下:
[0034]
(1)本发明α-咔啉的制备方法使用价格低廉的邻二氯苯、2-氨基吡啶为原料,在双金属催化下经c-n偶联和c-c偶联串联反应得到产品α-咔啉,反应过程中邻二氯苯既作为反应底物也作为反应溶剂,原料简单易得、反应条件温和、制备步骤简单,为合成α-咔啉提供了一种新的方法。
[0035]
(2)本发明采用镍/铜双金属协同催化,铜盐/草酰二胺催化体系促进c-n偶联反应进行生成相应的n-(2-氯苯基)吡啶-2-胺中间体,反应中间体中的吡啶和胺上的氮原子与铜盐配位形成环n-(2-氯苯基)吡啶-2-胺铜盐从而限制其以胺上氮原子为轴的自由旋转,使其吡啶和苯环的夹角减小,同时所形成的中间态产物中吡啶β位的反应活性增加,大大促进了吡啶β位上亲电取代反应,进而更有利于与镍催化剂生成环镍中间体,随后发生还原消除生成目标产物;若采用单一镍或铜盐催化剂,反应基本无法进行,只铜盐作为催化剂仅能促进第一步c-n偶联进行,无法催化随后进行的c-h/c-x偶联反应,只以镍盐为催化剂有可能发生发生第一步的c-n偶联反应,但是因为竞争关系n-(2-氯苯基)吡啶-2-胺可能进一步与另一分子的2-氨基吡啶反应,大大降低了反应的选择性,几乎无产物生成。
[0036]
(3)本发明与现有合成方法相比,本发明采用镍/铜双金属催化,避免贵金属催化剂的使用,大大降低了产品制备成本和提出难度,同时副产物环境危害小,反应选择性高、产品总收率高的特点,后处理提纯无需柱层析过程,具有生产成本低、合成步骤经济性、环境友好性的特点。
[0037]
(4)本发明的制备方法中邻二氯苯过量,反应过程中邻二氯苯既作反应底物又作反应溶剂,由于邻二氯苯的沸点为179℃,产品α-咔啉沸点为373℃,通过减压蒸馏可回收未反应的邻二氯苯,操作简单,绿色环保;反应过程中加入其他有机溶剂(例如邻二甲苯)反应基本无法进行,加入其他有机溶剂降低了邻二氯苯和2-氨基吡啶的反应浓度,使得反应很难进行,同时另加反应溶剂会与反应过程中生成的水发生共沸,从而降低反应温度,降低产品的溶解性使反应无法正向进行。
[0038]
(5)本发明α-咔啉的制备方法具有生产成本低、反应选择性好、产品收率高(可达95%以上)、环境友好的优势,有利于实施大规模生产,具备工业产业化可行性,拓展了α-咔啉类衍生物应用在医药等领域材料设计的合成方法。
具体实施方式
[0039]
以下通过具体实施例说明本发明的技术方案。应该理解,本发明提到的一个或者多个步骤不排斥在组合步骤前后还存在其他方法和步骤,或者这些明确提及的步骤间还可以插入其他方法和步骤。还应理解,这些实例仅用于说明本发明而不用于限制本发明的范围。除非另有说明,各方法步骤的编号仅为鉴别各方法步骤的目的,而非限制每个方法的排列次序或限定本发明的实施范围,其相对关系的改变或调整,在无实质技术内容变更的条件下,亦可视为本发明可实施的范畴。
[0040]
实施例中所采用的原料和仪器,对其来源没有特定限制,在市场购买或者按照本领域内技术人员熟知的常规方法制备即可。
[0041]
实施例1:一种α-咔啉的制备方法
[0042]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向500ml反应瓶内加入294.1g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的氧化亚铜(99%,10.0mmol)、1.07g的[ni(ipr*)cpcl](99%,1.0mmol)、2.7g的草酰二胺配体l1(99%,10.0mmol)、224.4g的叔丁醇钾(99%,2.0mol)和51.4g的溴化钠(99%,0.5mol);投料毕,升温至120℃,搅拌转速500rpm,保温反应24小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温,加入水和四氢呋喃萃取,有机层脱除溶剂经甲苯结晶得到143.9g的α-咔啉,收率
85.6%。
[0043]
实施例2:一种α-咔啉的制备方法
[0044]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向500ml反应瓶内加入294.1g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.90g的碘化亚铜(99%,10.0mmol)、1.19g的[ni(ipr*
tol
)cpcl](99%,1.0mmol)、3.19g的草酰二胺配体l2(99%,10.0mmol)、202.4g的三乙胺(99%,2.0mol)、119.0g的溴化钾(99%,1.0mol);投料毕,升温至130℃,搅拌转速500rpm,保温反应18小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层脱除溶剂经邻二甲苯结晶得到155.2g的α-咔啉,收率92.3%。
[0045]
实施例3:一种α-咔啉的制备方法
[0046]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向500ml反应瓶内加入220.6g的邻二氯苯(99%,1.5mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的溴化亚铜(99%,10.0mmol)、5.50g的[ni(ipr*
ome
)cpcl](99%,5.0mmol)、3.19g的草酰二胺配体l3(99%,10.0mmol)、456.72g的dbu(99%,3.0mol),150.0g的碘化钠(99%,1.0mol);投料毕,升温至90℃,搅拌转速500rpm,保温反应36小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层脱除溶剂经乙醇结晶得到161.1g的α-咔啉,收率95.8%。
[0047]
实施例4:一种α-咔啉的制备方法
[0048]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向1000ml反应瓶内加入735.6g的邻二氯苯(99%,5.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、0.99g的氯化亚铜(99%,10.0mmol)、1.19g的[ni(ipr*
tol
)cpcl](99%,1.0mmol)、3.19g的草酰二胺配体l4(99%,10.0mmol)、160.1g的叔丁醇锂(99%,2.0mol)、17.37g的溴化锂(99%,0.5mol);投料毕,升温至110℃,搅拌转速500rpm,保温反应20小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层脱除溶剂经乙醇结晶得到141.8g的α-咔啉,收率84.3%。
[0049]
实施例5:一种α-咔啉的制备方法
[0050]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向1000ml反应瓶内加入441.2g的邻二氯苯(99%,3.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的氧化亚铜(99%,10.0mmol)、1.10g的[ni(ipr*
ome
)cpcl](99%,1.0mmol)、3.19g的草酰二胺配体l3(99%,10.0mmol)、160.1g的叔丁醇锂(99%,2.0mol)、30.0g的碘化钠(99%,0.2mol);投料毕,升温至110℃,搅拌转速500rpm,保温反应24小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层脱除溶剂经乙醇结晶得到150.8g的α-咔啉,收率89.6%。
[0051]
实施例6:一种α-咔啉的制备方法
[0052]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向500ml反应瓶内加入294.2g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、0.143g的氧化亚铜(99%,1.0mmol)、1.07g的[ni(ipr*)cpcl](99%,1.0mmol)、0.638g的草酰二胺配体l2(99%,2.0mmol)、456.72g的dbu(99%,3.0mol)、51.4g的溴化钠(99%,0.5mol);投料毕,升温至130℃,搅拌转速500rpm,保温反应36小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层脱除溶剂经乙醇结晶得到140.9g的α-咔啉,收率
83.8%。
[0053]
实施例7:一种α-咔啉的制备方法
[0054]
一种α-咔啉的制备方法,步骤包括:在氮气保护下,向1000ml反应瓶内加入735.6g的邻二氯苯(99%,5.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的氧化亚铜(99%,10.0mmol)、0.12g的[ni(ipr*
tol
)cpcl](99%,0.1mmol)、2.7g的草酰二胺配体l1(99%,10.0mmol)、288.3g的叔丁醇钠(99%,3.0mol)、74.9g的碘化钠(99%,0.5mol);投料毕,升温至130℃,搅拌转速500rpm,保温反应36小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层脱除溶剂经乙醇结晶得到135.9g的α-咔啉,收率80.8%。
[0055]
对比例1:未加入配体
[0056]
本对比例与实施例1反应条件和参数相比,唯一不同在于对比例1未加入配体l1。
[0057]
在氮气保护下,向500ml反应瓶内加入294.1g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的氧化亚铜(99%,10.0mmol)、1.07g的[ni(ipr*)cpcl](99%,1.0mmol)、224.4g的叔丁醇钾(99%,2.0mol)、51.4g的溴化钠(99%,0.5mol);投料毕,升温至120℃,搅拌转速500rpm,保温反应24小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层取样经gc-ms分析,α-咔啉的定量收率<5%;本对比例的收率极低。
[0058]
对比例1与实施例1对比可知,本发明中草酰二胺配体对于反应至关重要,采用的草酰二胺配体是针对反应体系特别选择的特定类型的配体,通过改变配体吡啶环上取代基位置和吡啶环个数增强配体的配位能力及空间位阻,从而增强了与铜盐的配位能力,促进铜盐催化邻二氯苯与2-氨基吡啶发生c-n偶联反应,催化效果明显提高;反应过程中不加草酰二胺配体,反应基本无法进行。
[0059]
对比例2:另加入溶剂
[0060]
本对比例与实施例1反应条件和参数相比,唯一不同在于对比例2邻二甲苯作为反应溶剂。
[0061]
在氮气保护下,向500ml反应瓶内加入294.1g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的氧化亚铜(99%,10.0mmol)、1.07g的[ni(ipr*)cpcl](99%,1.0mmol)、2.7g的草酰二胺配体l1(99%,10.0mmol)、224.4g的叔丁醇钾(99%,2.0mol)、51.4g的溴化钠(99%,0.5mol)、150ml的邻二甲苯;投料毕,升温至120℃,搅拌转速500rpm,保温反应24小时;反应结束后,减压回收未反应的邻二氯苯和反应溶剂邻二甲苯,冷却室温加入水和四氢呋喃萃取,有机层取样经gc-ms分析,α-咔啉的定量收率<5%;本对比例的收率极低。
[0062]
对比例2与实施例1对比可知,另加反应溶剂会阻碍反应的进行,这是由于一方面加入其他有机溶剂降低了邻二氯苯和2-氨基吡啶的反应浓度,使得反应很难进行,同时另加反应溶剂会与反应过程中生成的水发生共沸,从而降低反应温度,降低产品的溶解性使反应无法正向进行。
[0063]
对比例3:单一铜催化剂
[0064]
本对比例与实施例1反应条件和参数相比,唯一不同在于对比例3催化剂仅使用氧化亚铜。
[0065]
在氮气保护下,向500ml反应瓶内加入294.1g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol)、1.43g的氧化亚铜(99%,10.0mmol)、2.7g的草酰二胺配体l1(99%,10.0mmol)、224.4g的叔丁醇钾(99%,2.0mol)、51.4g的溴化钠(99%,0.5mol);投料毕,升温至120℃,搅拌转速500rpm,保温反应24小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层取样经gc-ms分析,未检测到产物α-咔啉生成。
[0066]
对比例4:单一镍催化剂
[0067]
本对比例与实施例1反应条件和参数相比,唯一不同在于对比例4催化剂仅使用[ni(ipr*)cpcl]。
[0068]
在氮气保护下,向500ml反应瓶内加入294.1g的邻二氯苯(99%,2.0mol)、94.1g的2-氨基吡啶(99%,1.0mol),1.07g的[ni(ipr*)cpcl](99%,1.0mmol)、2.7g的草酰二胺配体l1(99%,10.0mmol)、224.4g的叔丁醇钾(99%,2.0mol)、51.4g的溴化钠(99%,0.5mol);投料毕,升温至120℃,搅拌转速500rpm,保温反应24小时;反应结束后,减压回收未反应的邻二氯苯,冷却室温加入水和四氢呋喃萃取,有机层取样经gc-ms分析,未检测到产物α-咔啉生成。
[0069]
结合对比例3、对比例4,实施例1相比,可见若采用单一镍或铜盐催化剂,反应基本无法进行,只铜盐作为催化剂仅能促进第一步c-n偶联进行,无法催化随后进行的c-h/c-x偶联反应,只以镍盐为催化剂有可能发生发生第一步的c-n偶联反应,但是因为竞争关系n-(2-氯苯基)吡啶-2-胺可能进一步与另一分子的2-氨基吡啶反应,大大降低了反应的选择性,几乎无产物生成。采用镍/铜双金属协同催化,铜盐/草酰二胺催化体系促进c-n偶联反应进行生成相应的n-(2-氯苯基)吡啶-2-胺中间体,反应中间体中的吡啶和胺上的氮原子与铜盐配位形成环n-(2-氯苯基)吡啶-2-胺铜盐从而限制其以胺上氮原子为轴的自由旋转,使其吡啶和苯环的夹角减小,同时所形成的中间态产物中吡啶β位的反应活性增加,大大促进了吡啶β位上亲电取代反应,进而更有利于与镍催化剂生成环镍中间体,随后发生还原消除生成目标产物。
[0070]
前述对本发明的具体示例性实施方案的描述是为了说明和例证的目的。这些描述并非想将本发明限定为所公开的精确形式,并且很显然,根据上述教导,可以进行很多改变和变化。对示例性实施例进行选择和描述的目的在于解释本发明的特定原理及其实际应用,从而使得本领域的技术人员能够实现并利用本发明的各种不同的示例性实施方案以及各种不同的选择和改变。本发明的范围意在由权利要求书及其等同形式所限定。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。