水溶性ib族贵金属亚10纳米胶粒、其制备方法及应用
技术领域
1.本发明涉及一种纳米颗粒,尤其涉及一种水溶性ib族贵金属亚10纳米胶粒、其制备方法及其应用,属于贵金属纳米材料技术领域。
背景技术:
2.贵金属的使用可以追溯到人类历史早期发展阶段,并一直被视为优越的权力和财富的标志。随着工业文明的出现,贵金属坚固的性质,耐极端腐蚀和氧化条件,它们已被广泛应用于航空航天、汽车、化学、能源、电气和电子工业。21世纪以后,纳米技术的发展为贵金属材料提供了无数的可能性,人们发现和研究了贵金属材料在纳米尺度、团簇和单个原子上的光学、催化、电子和生物医学特性。这些独特的特性在催化、传感、能量转换、光子学和生物医学等领域具有巨大的应用潜力。
3.在催化方面,贵金属纳米颗粒在气相加氢/氧化、碳活化、氧化、偶联、还原和水裂解等反应中发挥了关键作用,基于纳米颗粒优异的催化性能,在化工生产当中得到了广泛应用,成为不可替代的重要一环。然而贵金属性质很大程度上取决于其物理化学特征,包括大小、形状、实体或空心内部和表面组成等。纳米颗粒不同的比表面积,颗粒尺寸,表面组成以及暴露晶面等属性强烈影响催化活性和选择性。此外,在双金属或多组分纳米结构的情况下,性能也取决于元素在粒子内的分布(合金或核-壳形态)。因此,通过调整和控制这些物理和化学参数,可以改变和优化目标应用所需的特征属性。
4.随着纳米技术的不断突破,现如今可以设计合成不同的形貌、尺寸、成分及结构贵金属纳米颗粒,但是面对工业生产的大量需求,实验室微量且昂贵的制备手段是贵金属纳米材料应用推广和普及的瓶颈,因此寻求一种简便的合成方法来制备具有高稳定性和催化专一性的低成本金属纳米颗粒催化剂是化工生产中的重要课题。然而,纳米颗粒的合成工业对合成步骤有严格的要求,通常要求以简单和合理的方式(很少的工艺步骤)进行合成,使用尽可能少的原材料,避免产生废料,而且在最终产品中痕量的污染物离子或分子对于特定的应用要求无任何影响。
5.一般来说,纳米颗粒的制备方法包含物理方法和化学方法,由于化学方法具有高效简单、成本低廉和精确可控等优势,成为纳米材料制备的主要方法。基于溶液的化学合成方法,主要包括盐前驱体在配体和稳定剂的存在下的还原或分解,其中配体起到了关键的作用,不仅可以调控纳米颗粒的尺寸、形貌及晶面,还起到了稳定纳米颗粒的作用。目前对于贵金属纳米颗粒的合成通常为单一的有机配体,例如柠檬酸根配体、多元醇配体、油胺配体、硫醇配体等,通过形成一系列的有机金属配合物,在一定的条件下经过聚集、熟化、生长等步骤形成纳米颗粒。基于该液相合成法已经在合成各种大小、形状和成分可控的贵金属纳米颗粒方面取得了相当大的成功(chemical society reviews,2018,47(14):5187-5233;acs nano,2015,9(7):7052-7071)。
6.但是液相合成法依然存在诸多弊端,比如合成过程通常在稀释的介质中进行,产生低纳米颗粒浓度的悬浮液。因此,对于许多实际应用,有必要通过沉淀和(或)离心来放大
和浓缩悬浮液,然而必要的后处理也可能引起纳米颗粒的变化或聚集,还需要纯化以除去过量的表面活性剂和未反应的前驱体。此外,纳米颗粒的溶液合成对反应条件的变化和溶液或溶剂中的痕量离子非常敏感,而且多数的液相反应需要高温环境,这会导致纳米颗粒合成的重复性和一致性难以控制。尽管最近使用流动条件取得了进展(chemicalengineering science,2018,189:422-430),可以在相对较低的温度稳定合成贵金属纳米颗粒,但在批量条件下溶液相合成配方的放大仍然有限。
7.在各种溶液法合成纳米颗粒的方法中,只有少数方法,可以很容易地放大合成。对于基于有机金属配合物的纳米颗粒的制备,由于反应条件温和,有机金属途径提供了很好的粒子形成过程的控制。此外,由于金属配合物的性质,不引入污染物,如卤化物或其他离子。然而,有机金属配合物难以制备,成本昂贵,因为它们固有的分解倾向,通常需要在惰性气氛中处理。其他几种制备方法依赖于使用介质到强还原剂,如联氨、硼氢化钠或由伽玛射线辐照产生的溶剂化电子来还原金属前驱体。如果这些还原剂在低沸点溶剂中稀释,一般很难获得结晶良好的颗粒,需要随后的热处理。可以使用溶剂热条件,但消耗大量的能量。此外,为了获得非常低的尺寸分散性,这些还原反应可以在特定的条件下进行,如微乳液技术,但是用于合成的大量溶剂严重阻碍了大规模生产,然而这些配体要么只能存在于油相中,要么形成的纳米颗粒稳定性较低,同时表面配体吸附在贵金属纳米颗粒的表面难以去除,影响催化活性及其他应用。
8.针对以上问题,研究人员提出了除溶液法以外的多种化学方法来提高纳米颗粒的稳定性和催化活性。
9.基于微乳液的金属纳米粒子的制备已成为人们关注的热点,同时也成为一种能够更好地控制合成纳米粒子的尺寸和形状等物理特性的有效方法。一般来说,微乳液是两种不相溶的液体在表面活性剂存在下的简单混合物。这些体系通常具有超低的界面张力、较大的界面面积和热力学稳定性。
10.电化学方法于1994年由reetz和helbig首次提出,他们将阳极上的纯金属片溶解,在电解液存在的情况下在电化学电池的阴极上沉积金属盐,从而产生纳米颗粒,电化学工艺制备的贵金属纳米颗粒主要因其催化性能而被应用,最近已作为生物传感器应用于生物医学领域(russian chemical reviews,2018,87(11):1080.)。
11.此外,微波辅助合成已经快速发展成为一种高可靠、快速和简单的方法,且支持纳米颗粒的形态控制。在无溶剂或表面活性剂的条件下,微波诱导放电也可用于从金属颗粒中合成cu、ni、zn纳米粒子(beilsteinjournal ofnanotechnology,2020,11(1):1019-1025.)。该方法基于偶极子相互作用的原理(分子倾向于排列自己,并随着微波振荡电场的步调振荡,它们之间的碰撞和摩擦会产生热量)和离子传导(电场产生离子运动,因为分子试图向快速变化的场定向,导致瞬时超热),产生加热效应,导致金属离子还原为纳米颗粒。
12.对经济和环境友好方法发展的追求使人们着眼于用植物提取物或微生物作为纳米颗粒合成的重要手段。其中生物系统已被研究人员用作纳米材料制备的细胞工厂,原核生物(细菌)和真核生物(藻类、真菌和植物)都被用于绿色合成纳米颗粒的方法(journal ofnanobiotechnology,2018,16(1):1-28.)。
13.上述通过化学方法合成的贵金属纳米颗粒的稳定机理可通过经典的dlvo理论进行阐释。在纳米颗粒溶液中,带正电或负电的表面配体会使纳米粒子以其特有的双电层结
构存在,其最外层的扩散层离子会将纳米粒子里层所带的电荷中和,以保持整个粒子呈中性。因此,粒子的扩散层就如同一个具有屏蔽作用的“屏蔽层”。然而,这种双电层很容易被破环,当两个粒子不断靠近,其自身的扩散层就会接近并发生重叠现象,电荷分布受到了影响和破坏,产生了静电斥力,打破了原有的稳定存,从而相互聚集融合形成大颗粒。
技术实现要素:
14.本发明的主要目的在于提供一种双配体稳定的水溶性ib族贵金属亚10纳米胶粒及其制备方法,克服现有的贵金属纳米无法兼顾溶液稳定性与溶度的不足,同时该方法满足可规模化制备的要求。
15.本发明的另一目的还在于提供所述水溶性ib族贵金属亚10纳米胶粒的应用。
16.为实现前述发明目的,本发明采用的技术方案包括:
17.本发明实施例提供了一种水溶性ib族贵金属亚10纳米胶粒具有核壳结构,其包括作为核的ib族贵金属纳米颗粒,以及分布于ib族贵金属核表面的壳层,所述壳层为双层结构,所述双层结构包括内层的过渡金属阳离子,以及外层的有机配体阴离子,所述ib族贵金属纳米颗粒的尺寸小于10纳米。
18.本发明实施例还提供了一种水溶性ib族贵金属亚10纳米胶粒的制备方法,其包括:
19.使有机配体水溶液与ib族贵金属盐水溶液反应,制得前驱体溶液;
20.将添加剂加入到前驱体溶液中,制得反应体系;
21.将还原剂缓慢滴加到所述反应体系中,于25~75℃反应0.5~3h,制得固体纳米颗粒;
22.将所述固体纳米颗粒与稳定剂混合,得到水溶性ib族贵金属亚10纳米胶粒。
23.在一些实施例中,所述水溶性ib族贵金属亚10纳米胶粒的制备方法包括:
24.将ib族贵金属盐在无氧条件下溶解,制得ib族贵金属盐水溶液;
25.将可溶性过渡金属盐溶解在水中,制得可溶性过渡金属盐水溶液,作为还原剂;
26.将水溶性有机配体溶解在水中,制得有机配体水溶液;
27.将添加剂溶解在水中,制得添加剂水溶液;
28.将所述有机配体水溶液加入到ib族贵金属盐水溶液中,升温至25~70℃,制得前驱体溶液;
29.将所述添加剂溶液加入到前驱体溶液中,制得反应体系;
30.将所述还原剂缓慢滴加到所述反应体系中反应,反应温度为25~75℃,反应时间为0.5~3h,制得固体纳米颗粒;
31.将所述固体纳米颗粒与稳定剂混合,得到水溶性ib族贵金属亚10纳米胶粒溶液。
32.本发明实施例还提供了所述水溶性ib族贵金属亚10纳米胶粒于制备催化剂、导电浆料、印刷油墨或纳米流体等材料中的应用。
33.与现有技术相比,本发明的有益效果包括:
34.1)本发明制备的水溶性ib族贵金属亚10纳米胶粒具有较高的抗离子污染能力和热稳定性;
35.2)本发明制备的水溶性ib族贵金属亚10纳米胶粒可得到较高浓度的水溶液,浓缩
液最高可达100g/l,可将浓缩液稀释至任意符合使用要求的浓度;
36.3)本发明制备的水溶性ib族贵金属亚10纳米胶粒单位质量浓度制备所需原材料投入较少,操作简便易用实施,易于存储运输和规模化生产;
37.4)本发明还提供了水溶性ib族贵金属亚10纳米胶粒的新应用导向,所述纳米胶粒可适用于多种化工生产中的催化剂,也可用于水性导电浆料、印刷油墨、纳流体等材料的制备。
附图说明
38.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明中记载的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
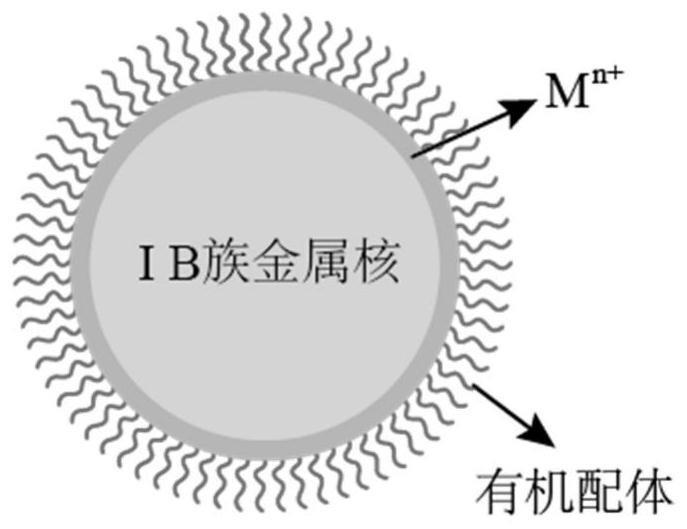
39.图1为本发明典型实施方案中水溶性ib族贵金属亚10纳米胶粒的结构示意图。
40.图2为本发明实施例1中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
41.图3为本发明实施例2中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
42.图4为本发明对比例2中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
43.图5为本发明实施例3中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
44.图6为本发明实施例4中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
45.图7为本发明实施例5中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
46.图8为本发明实施例6中提供的水溶性ib族贵金属亚10纳米胶粒的tem图。
具体实施方式
47.鉴于现有技术的不足,本案发明人经长期研究和大量实践,提出了本发明的设计思路与方案,其主要是提出一种水溶性ib族贵金属亚10纳米胶粒。
48.本发明的主要发明构思在于:提出一种通过具有还原性的低价态过渡金属离子还原贵金属前驱体,在相应配体和稳定剂的作用下得到纳米胶粒。首先,将贵金属盐、过渡金属盐、水溶性有机配体和添加剂在无氧条件下进行溶剂化处理,然后将溶剂化的过渡金属离子和贵金属离子在原子级别进行混合,由于电负性的差异导致金属离子之间会发生电子转移,被还原的贵金属离子在溶液中形成稳定晶核,于此同时在晶核生长的驱动力作用下过渡金属离子扩散至晶核表面,形成纳米颗粒壳层。与传统还原剂相比,所得纳米颗粒表面形成一种特殊的过渡金属保护层,表面为双层结构的纳米颗粒,内层为过渡金属阳离子,外层为有机配体阴离子。纳米胶粒表面由于过渡金属阳离子的存在,增大了纳米颗粒之间的势垒,可避免纳米颗粒因高表面能导致的聚合生长,从而表现出更强的稳定性。与此同时,该方法反应条件温和,反应体系简单,易于规模化生产。
49.请参阅图1所示,本发明实施例的一个方面提供的一种双层配体稳定的水溶性ib族贵金属亚10纳米胶粒(即纳米粒子结构)具有核壳结构,包括作为核的ib族贵金属纳米颗粒,以及分布于ib族贵金属核表面的壳层,所述壳层为双层结构,所述纳米颗粒表面的双层结构中内层为过渡金属阳离子,外层为有机配体阴离子,所述ib族贵金属纳米颗粒的尺寸小于10纳米。
50.在一些实施方案中,所述核的尺寸范围为3~10nm,所述壳层的厚度范围为0.5~1nm。
51.进一步地,所述内层的厚度范围为0.5~1nm。
52.在一些实施方案中,所述ib族贵金属纳米颗粒包括au、ag、cu等中的任意一种或至少两种组成的合金。
53.在一些实施方案中,所述过渡金属阳离子包括亚铜离子、亚钒离子、亚铟离子、亚锡离子、亚铁离子、亚铬离子等中的任意一种或两种以上的组合,但不限于此。
54.在一些实施方案中,所述有机配体阴离子来源于有机配体,所述有机配体包括聚乙烯吡咯烷酮、柠檬酸钠、聚丙烯酸、单宁酸、硫辛酸等中的任意一种或两种以上的组合,但不限于此。
55.本发明实施例的另一个方面提供的一种水溶性ib族贵金属亚10纳米胶粒(下文亦可称为“水溶性i b族贵金属亚10纳米颗粒”)的制备方法主要包括以下三个步骤,分别为反应液的配制、贵金属离子的还原和纳米颗粒的稳定。具体包括:
56.使有机配体水溶液与ib族贵金属盐水溶液反应,制得前驱体溶液;
57.将添加剂加入到前驱体溶液中,制得反应体系;
58.将还原剂缓慢滴加到所述反应体系中,于25~75℃反应0.5~3h,制得固体纳米颗粒;
59.将所述固体纳米颗粒与稳定剂混合,得到水溶性ib族贵金属亚10纳米胶粒。
60.在一些优选实施方案中,所述制备方法具体包括:
61.将ib族贵金属盐在无氧条件下溶解,制得ib族贵金属盐水溶液;
62.将可溶性过渡金属盐溶解在水中,制得可溶性过渡金属盐水溶液,作为还原剂;
63.将水溶性有机配体溶解在水中,制得有机配体水溶液;
64.将添加剂溶解在水中,制得添加剂水溶液;
65.将所述有机配体水溶液加入到ib族贵金属盐水溶液中,升温至25~70℃,制得前驱体溶液;
66.将所述添加剂溶液加入到前驱体溶液中,制得反应体系;
67.将所述还原剂缓慢滴加到所述反应体系中反应,反应温度为25~75℃,反应时间为0.5~3h,制得固体纳米颗粒;
68.将所述固体纳米颗粒与稳定剂混合,得到水溶性ib族贵金属亚10纳米胶粒溶液。
69.其中,在一些更为具体的实施方案之中,所述水溶性ib族贵金属亚10纳米胶粒溶液的制备方法包括如下步骤:
70.(1)将ib族贵金属盐在无氧条件下溶解,制备ib族贵金属盐水溶液,即“金属盐水溶液a”;
71.(2)将一定量的可溶性过渡金属盐溶解在去离子水中,制得可溶性过渡金属盐水溶液,即“还原剂溶液b”;
72.(3)将一定量的水溶性有机配体溶解在去离子水中,制得有机配体水溶液,即“配体溶液c”;
73.(4)将一定量的水溶性添加剂或稳定剂溶解在去离子水中,制得添加剂水溶液,即“添加剂溶液d”;
74.(5)将配体溶液c加入到金属盐水溶液a中,缓慢升温至25~70℃,制得前驱体e;
75.(6)将添加剂溶液d加入到前驱体溶液e中,制得反应液f;
76.(7)将还原剂溶液b缓慢滴加反应液f中,反应温度为25~75℃,反应时间为0.5~3h;
77.(8)反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒;
78.(9)将所得固体纳米颗粒和适当的稳定剂混合,得到水溶性的ib族贵金属亚10纳米颗粒溶液。
79.在一些实施例中,所述水溶性ib族贵金属亚10纳米胶粒溶液中贵金属的浓度为0.1~100g/l。
80.在一些实施例中,所述水溶性的i b族贵金属亚10纳米颗粒溶液以质量分数计包含i b族贵金属0.1~100g/l;过渡金属阳离子0.5~70g/l;有机配体阴离子0.1~0.5kg/l;其他稳定剂0.1~30g/l;其他添加剂0.1~20g/l。
81.在一些实施例中,所述ib族贵金属为au、ag、cu中的一种或至少其中两种的组合。
82.在一些实施例中,步骤(1)中,所述ib族贵金属盐包括铜盐、银盐、金盐中的任意一种或两种以上的组合。
83.进一步地,所述ib族贵金属盐包括cu(no3)2、cucl2、agno3、ch3cooag、haucl3等中的任意一种或两种以上的组合,但不限于此。
84.在一些实施例中,步骤(2)中,所述可溶性过渡金属盐包括亚铜类盐、亚钒类盐、亚铟类盐、亚锡盐类盐、亚铁盐类盐、亚铬类盐等中的任意一种或两种以上的组合,但不限于此。
85.进一步地,所述水溶性i b族贵金属亚10纳米胶粒溶液中可溶性过渡金属盐的浓度为0.5~70g/l。
86.进一步地,所述可溶性过渡金属盐包括cucl、ch3coocu、cu2so4、voso4、vo2no3、incl、feso4、fecl2、fe(no3)2、fe(ch3coo)2、fe2o7p2、snso4、sn(ch3coo)2、sn2o7p2、sncl2、crcl2、c4h8cro5等中的任意一种或两种以上的组合,但不限于此。
87.在一些实施例中,步骤(3)中,所述水溶性有机配体包括聚乙烯吡咯烷酮k30、柠檬酸钠、聚丙烯酸、单宁酸、硫辛酸等中的任意一种或两种以上的组合,但不限于此。
88.进一步地,所述水溶性ib族贵金属亚10纳米胶粒溶液中水溶性有机配体的浓度为0.1~0.5kg/l。
89.在一些实施例中,步骤(4)中,所述添加剂为两性化合物,优选自氨基乙酸、甜菜碱、甘氨酸、乙二胺四乙酸、羟乙基乙二胺三乙酸、二亚乙基三胺五乙酸、二氨基丙酸、谷氨酸等中的任意一种或两种以上的组合,但不限于此。
90.进一步地,所述添加剂水溶液中添加剂的浓度为0.1~20g/l。
91.在一些实施例中,步骤(9)中,所述稳定剂为水溶性醇类有机物,优选包括丙三醇、乙二醇、聚乙二醇、聚乙烯醇等中的任意一种或两种以上的组合,但不限于此。
92.进一步地,所述水溶性ib族贵金属亚10纳米胶粒溶液中稳定剂的浓度为0.1~30g/l。
93.在一些实施例中,步骤(9)中,所述固体纳米颗粒与稳定剂的质量比为1∶0.5~1∶2。
94.在一些实施例中,所述水溶性有机配体的摩尔量为2an~6an,其中a代表ib族贵金属的摩尔量,n代表ib族贵金属的电荷数。
95.在一些实施例中,所述可溶性过渡金属盐所含过渡金属阳离子的摩尔量为a(n 1)/m~3a(n 1)/m,其中a代表ib族贵金属的摩尔量,n代表ib族贵金属的电荷数,m代表过渡金属阳离子反应前后得失电子数。
96.在一些实施例中,所述添加剂或稳定剂的摩尔量为an~8an,其中a代表ib族贵金属的摩尔量,n代表ib族贵金属的电荷数。
97.综上所述,本发明制备的水溶性ib族贵金属亚10纳米胶粒具有较高的抗离子污染能力和热稳定性。并且,本发明制备的水溶性ib族贵金属亚10纳米胶粒可得到较高浓度的水溶液,浓缩液最高可达100g/l,可将浓缩液稀释至任意符合使用要求的浓度。同时,本发明制备的水溶性ib族贵金属亚10纳米胶粒单位质量浓度制备所需原材料投入较少,操作简便易用实施,易于存储运输和规模化生产。
98.本发明实施例的另一个方面还提供了所述水溶性ib族贵金属亚10纳米胶粒的应用。具体的,所述纳米胶粒可用于多种化工生产中的催化剂,也可用于导电浆料、印刷油墨、纳米流体等材料的制备。
99.下面结合具体实施例和说明书附图对本发明的技术方案进行详细说明,所描述实例并不仅限于此,且基于本发明的实例本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。下面所用的实施例中所采用的实验材料,如无特殊说明,均可由常规的生化试剂公司购买得到。
100.实施例1
101.水溶性ib族贵金属亚10纳米胶粒的制备包含以下步骤:
102.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和硝酸铜加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述硝酸银和硝酸铜的质量比为1∶0.9,所述硝酸银的质量为8.49g。
103.步骤(2):配制还原剂溶液,称取一定量的硫酸氧钒加入到20ml去离子水中,充分搅拌至完全溶解。所述硫酸氧钒与贵金属的质量之比为1.2∶1。
104.步骤(3):配制配体溶液,称取一定量的柠檬酸钠加入到20ml去离子水中,充分搅拌至完全溶解。所述柠檬酸钠与贵金属的质量之比为5.4∶1。
105.步骤(4):配制添加剂溶液,称取一定量的甘氨酸加入到10ml去离子水中,分搅拌至完全溶解。所述甘氨酸与贵金属的质量之比为1∶1。
106.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至55℃,制得前驱体溶液。
107.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
108.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为55℃,反应时间为1小时。
109.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
110.步骤(9):将所得固体纳米颗粒和丙三醇按照质量比为1∶1混合,得到水溶性i b族
贵金属亚10纳米胶粒溶液。
111.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图2为本实施例1制得的纳米胶粒的tem照片,平均粒径为4.19nm。
112.制备过程中贵金属的质量浓度范围在0.1~100g/l之间可调,相应的配体、还原剂、添加剂应等比例缩放,以下实施例和对比例同样适用。
113.对比例1
114.水溶性i b族贵金属亚10纳米胶粒的制备包含以下步骤:
115.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和硝酸铜加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述硝酸银和硝酸铜的质量比为1∶0.9,所述硝酸银的质量为8.49g。
116.步骤(2):配制还原剂溶液,称取一定量的硫酸氧钒加入到20ml去离子水中,充分搅拌至完全溶解。所述硫酸氧钒与贵金属的质量之比为1.2∶1。
117.步骤(3):配制配体溶液,称取一定量的柠檬酸钠加入到20ml去离子水中,充分搅拌至完全溶解。所述柠檬酸钠与贵金属的质量之比为5.4∶1。
118.步骤(4):配制添加剂溶液,称取一定量的甘氨酸加入到10ml去离子水中,分搅拌至完全溶解。所述甘氨酸与贵金属的质量之比为1∶1。
119.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至55℃,制得前驱体溶液。
120.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
121.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为55℃,反应时间为1小时。
122.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
123.步骤(9):将所得固体纳米颗粒用适量的水分散,得到水溶性ib族贵金属亚10纳米胶粒溶液。
124.对比例1与实施例1相比,由于未在离心分离后添加适量的丙三醇分散,而是用水分散,导致所得水溶性i b族贵金属亚10纳米胶粒溶液在室温放置3天以后可以明显观察到溶液底部有沉淀。这是由于纳米尺度的贵金属纳米颗粒具有的高表面能,通过聚沉的方式相互融合生长,达到总自由能最小的稳定状态。加入丙三醇等醇类溶剂之后可以显著减少聚沉的发生,维持纳米颗粒的稳定分散状态。
125.实施例2
126.水溶性i b族贵金属亚10纳米胶粒的制备包含以下步骤:
127.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和氯金酸加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述氯金酸和硝酸银的质量比为2∶1,所述硝酸银的质量为8.49g。
128.步骤(2):配制还原剂溶液,称取一定量的硫酸亚锡加入到20ml去离子水中,充分搅拌至完全溶解。所述硫酸亚锡与贵金属的质量之比为2.1∶1。
129.步骤(3):配制配体溶液,称取一定量的柠檬酸钠加入到20ml去离子水中,充分搅
拌至完全溶解。所述柠檬酸钠与贵金属的质量之比为5.4∶1。
130.步骤(4):配制添加剂溶液,称取一定量的甜菜碱加入到10ml去离子水中,分搅拌至完全溶解。所述甜菜碱与贵金属的质量之比为1∶1。
131.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至55℃,制得前驱体溶液。
132.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
133.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为55℃,反应时间为1小时。
134.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
135.步骤(9):将所得固体纳米颗粒和乙二醇按照质量比为1∶1混合,得到水溶性ib族贵金属亚10纳米颗粒溶液。
136.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图3为本实施例制得的纳米颗粒的tem照片,平均粒径为4.81nm。
137.对比例2
138.水溶性i b族贵金属亚10纳米胶粒的制备包含以下步骤:
139.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和氯金酸加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述氯金酸和硝酸银的质量比为2∶1,所述硝酸银的质量为8.49g。
140.步骤(2):配制还原剂溶液,称取一定量的硫酸亚锡加入到20ml去离子水中,充分搅拌至完全溶解。所述硫酸亚锡与贵金属的质量之比为2.1∶1。
141.步骤(3):配制配体溶液,称取一定量的柠檬酸钠加入到20ml去离子水中,充分搅拌至完全溶解。所述柠檬酸钠与贵金属的质量之比为5.4∶1。
142.步骤(4):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至55℃,制得前驱体溶液。
143.步骤(5):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
144.步骤(6):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为55℃,反应时间为1小时。
145.步骤(7):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
146.步骤(8):将所得固体纳米颗粒和乙二醇混合,得到水溶性i b族贵金属亚10纳米颗粒溶液。
147.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图4为对比例制得的纳米颗粒的tem照片,平均粒径为17.6nm。
148.对比例2与实施例2相比,由于未添加两性化合物添加剂,从图4的tem中可以看出纳米颗粒的尺寸显著变大。贵金属纳米颗粒的形成和稳定与纳米颗粒表面吸附电解质的能力有关,电解质的吸附会在纳米颗粒表面形成双电层,而溶液的ph值对电解质的吸附会产
生强烈的影响。水溶性两性化合物可以维持反应溶液中的酸碱平衡,保证纳米颗粒表面电荷稳定,从而形成颗粒尺寸均匀的纳米颗粒。
149.实施例3
150.水溶性i b族贵金属亚10纳米颗粒的制备包含以下步骤:
151.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和氯金酸加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述氯金酸和硝酸银的质量比为2∶1,所述硝酸银的质量为8.49g。
152.步骤(2):配制还原剂溶液,称取一定量的硫酸亚铜加入到20ml去离子水中,充分搅拌至完全溶解。所述硫酸亚铜与贵金属的质量之比为2.2∶1。
153.步骤(3):配制配体溶液,称取一定量的硫辛酸加入到20ml去离子水中,充分搅拌至完全溶解。所述硫辛酸与贵金属的质量之比为2.4∶1。
154.步骤(4):配制添加剂溶液,称取一定量的氨基乙酸加入到10ml去离子水中,分搅拌至完全溶解。所述氨基乙酸与贵金属的质量之比为1∶1。
155.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至70℃,制得前驱体溶液。
156.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
157.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为70℃,反应时间为1.5小时。
158.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
159.步骤(9):将所得固体纳米颗粒和丙三醇按照质量比为1∶0.5混合,得到水溶性i b族贵金属亚10纳米颗粒溶液。
160.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图5为本施例制得的纳米颗粒的tem照片,平均粒径为4.76nm。
161.实施例4
162.水溶性i b族贵金属亚10纳米颗粒的制备包含以下步骤:
163.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和氯金酸加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述氯金酸和硝酸银的质量比为2∶1,所述硝酸银的质量为8.49g。
164.步骤(2):配制还原剂溶液,称取一定量的焦磷酸亚锡加入到20ml去离子水中,充分搅拌至完全溶解。所述焦磷酸亚锡与贵金属的质量之比为4∶1。
165.步骤(3):配制配体溶液,称取一定量的pvpk30加入到20ml去离子水中,充分搅拌至完全溶解。所述pvp k30与贵金属的质量之比为3∶1。
166.步骤(4):配制添加剂溶液,称取一定量的乙二胺四乙酸加入到10ml去离子水中,分搅拌至完全溶解。所述乙二胺四乙酸与贵金属的质量之比为2∶1。
167.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至70℃,制得前驱体溶液。
168.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制
得中间体反应液。
169.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为75℃,反应时间为0.5小时。
170.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
171.步骤(9):将所得固体纳米颗粒和丙三醇按照质量比为1∶2混合,得到水溶性i b族贵金属亚10纳米颗粒溶液。
172.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图6为本施例制得的纳米颗粒的tem照片,平均粒径为5.06nm。
173.实施例5
174.水溶性i b族贵金属亚10纳米颗粒的制备包含以下步骤:
175.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将硝酸银和硝酸铜加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述硝酸银和硝酸铜的质量为1∶0.9,所述硝酸银的质量为8.49g。
176.步骤(2):配制还原剂溶液,称取一定量的氯化亚铬加入到20ml去离子水中,充分搅拌至完全溶解。所述氯化亚铬与贵金属的质量之比为1.9∶1。
177.步骤(3):配制配体溶液,称取一定量的pvpk30加入到20ml去离子水中,充分搅拌至完全溶解。所述pvp k30与贵金属的质量之比为3∶1。
178.步骤(4):配制添加剂溶液,称取一定量的乙二胺四乙酸加入到10ml去离子水中,分搅拌至完全溶解。所述乙二胺四乙酸与贵金属的质量之比为2∶1。
179.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至60℃,制得前驱体溶液。
180.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
181.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为60℃,反应时间为2.5小时。
182.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
183.步骤(9):将所得固体纳米颗粒和丙三醇按照质量比为1∶1混合,得到水溶性ib族贵金属亚10纳米颗粒溶液。
184.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图7为本施例制得的纳米颗粒的tem照片,平均粒径为3.89nm。
185.实施例6
186.水溶性i b族贵金属亚10纳米颗粒的制备包含以下步骤:
187.步骤(1):配制贵金属溶液,在三口烧瓶中添加50ml去离子水,然后通氮气30min,再将氯金酸和硝酸铜加入到去离子水中,搅拌至完全溶解得到澄清溶液,所述氯金酸和硝酸铜的质量比为2∶1,所述硝酸银的质量为8.49g。
188.步骤(2):配制还原剂溶液,称取一定量的氯化亚铟加入到20ml去离子水中,充分搅拌至完全溶解。所述氯化铟与贵金属的质量之比为1.8∶1。
189.步骤(3):配制配体溶液,称取一定量的柠檬酸三钠加入到20ml去离子水中,充分搅拌至完全溶解。所述柠檬酸三钠与贵金属的质量之比为5.4∶1。
190.步骤(4):配制添加剂溶液,称取一定量的乙二胺四乙酸加入到10ml去离子水中,分搅拌至完全溶解。所述乙二胺四乙酸与贵金属的质量之比为2∶1。
191.步骤(5):将步骤(3)的配体溶液加入到步骤(1)中的贵金属盐水溶液中,缓慢升温至25℃,制得前驱体溶液。
192.步骤(6):将步骤(4)的添加剂溶液加入到前驱体溶液步骤(5)的前驱体溶液中,制得中间体反应液。
193.步骤(7):将步骤(2)的还原剂溶液缓慢滴加至步骤(6)的中间体反应液中,反应温度为25℃,反应时间为3小时。
194.步骤(8):反应结束后将反应液收集进行离心分离,倒掉上清液,得固体纳米颗粒。离心转速为5000rpm,离心时间为5min。
195.步骤(9):将所得固体纳米颗粒和聚乙二醇按照质量比为1∶0.8混合,得到水溶性i b族贵金属亚10纳米颗粒溶液。
196.取少量样品用去离子水稀释至适当浓度,可观察到溶液为有色透明溶液,图8为本对比例制得的纳米颗粒的tem照片,平均粒径为4.03nm。
197.应用例1
198.实施例1-6中所得贵金属纳米颗粒用于进一步制备高导热贵金属纳米流体,该制备方法包括以下步骤:
199.步骤(1),将贵金属元素质量分数为20-100g/l的纳米颗粒浆料与基液混合,所述纳米颗粒浆料与基液的质量比为1∶2~1∶4,在45℃下机械搅拌1小时,搅拌速度为500rpm,得到混合溶液a。
200.步骤(2),将混合溶液a与表面活性剂混合,所述混合溶液与表面活性剂的质量比为1∶0.001-1∶0.005,在45℃下机械搅拌1小时,搅拌速度为500rpm,得到混合溶液b。
201.步骤(3),再将所得混合溶液b放置于超声波细胞粉碎机中,所述超声波细胞粉碎机功率为30kw,所述操作条件为25℃下超声1小时,得到高导热贵金属纳米流体。
202.所述步骤(1)中,基液的选自丁醇、丙醇、乙二醇、甲苯、甘油、导热油中的一种或两种以上。
203.所述步骤(2)中,表面活性剂选自十二烷基苯磺酸钠、十二烷基氨基丙酸钠、壬基酚聚氧乙烯醚、壬基酚聚氧乙烯醚、聚乙烯吡咯烷酮中的一种。
204.所述高导热贵金属纳米流体具有优异的性能,其冰点可以达到-60℃,沸点高达200℃,而100℃时导热系数可高达0.508w/mk以上。
205.应用例2
206.实施例1-6中所得贵金属纳米颗粒用于进一步制备低温烧结水性贵金属纳米导电油墨,该制备方法包含以下步骤:
207.步骤(1),将质量分数为20-100g/l的贵金属纳米颗粒浆料与功能助剂混合,在室温下机械搅拌1小时,搅拌速度为500rpm,得混合溶液a。
208.步骤(2),将所得混合溶液a放置于超声波细胞粉碎机中,所述超声波细胞粉碎机功率为30kw,所述操作条件为25℃下超声1小时,得到低温烧结水性贵金属纳米导电油墨。
209.所述步骤(1)中功能助剂包含缓冲剂、保湿剂、表面活性剂、消泡剂和粘结剂。
210.所述缓冲剂选自异丁醇胺、三羟甲基氨基甲烷、4-羟乙基哌嗪乙磺酸中的一种。
211.所述保湿剂选自乙二醇、丙三醇、聚乙烯醇、聚乙二醇、丙二醇甲基醚中的一种。
212.中所述表面活性剂选自杜邦氟碳表面活性剂fs-300、杜邦氟碳表面活性剂fsa、surfynol465、surfynol 104bc中的一种或两种。
213.所述消泡剂选自三丁基磷酸盐、有机硅消泡剂和正戊醇中的一种或多种。
214.所述粘结剂sma2000、mace 85-302-1、聚氨基甲酸酯、聚乙烯醇、聚乙烯吡咯烷酮、丙烯酸树脂、羧甲基纤维素钠和羧乙基纤维素钠中的一种或多种。
215.所述水性贵金属纳米导电油墨可用于丝网印刷、直写工艺和喷墨打印。
216.应用例3
217.本发明提供了水溶性i b族贵金属亚10纳米颗粒的工业催化实用实例。用于非金属材料表面化学镀铜的催化过程,表现出具有较好的催化效果,与现有的技术相比,本发明提供的贵金属纳米催化剂是一种中性的水溶液溶剂,具有较好的稳定性、可靠性和环境友好性。具体包含以下操作步骤:
218.步骤(1),准备一块树脂材料基材,例如:abs树脂、pcb板、芳纶树脂、聚酰亚胺树脂等。
219.步骤(2),将所述材料进行表面粗化处理,增强化学镀铜附着力。
220.步骤(3),将步骤(2)中处理过的树脂材料基材进行表面改性处理,调整表面电荷。
221.步骤(4),将步骤(3)中处理过的树脂材料基材进行活化处理(表面吸附贵金属纳米颗粒),所述活化液为实施例1和对比例1-5中所得贵金属纳米颗粒。将水溶性i b族贵金属亚10纳米颗粒用溶剂稀释至200-1000ppm,所述溶剂为去离子水。
222.步骤(5),将步骤(4)中处理过的树脂材料基材浸入经典化学镀铜液中,处理完后得到表面镀铜的树脂材料,表1为通过本实施例的纳米颗粒催化得到的化学镀铜效果。
223.表1 ib族贵金属纳米颗粒的催化镀铜效果
224.实施例催化剂稳定性起镀时间化学镀效果实施例160℃,72h103s
○○○○
实施例260℃,48h127s
○○○
实施例360℃,24h69s
○○○○○
实施例460℃,72h72s
○○○○
实施例560℃,36h101s
○○○○
实施例660℃,72h110s
○○○○○
225.此外,本案发明人还利用前文所列出的其它原料以及其它工艺条件等替代实施例1中的各种原料及相应工艺条件进行了相应试验,所需要验证的内容和与实施例1产品均接近。
226.应当理解,以上说明及在图纸上所示的实施例,不可解析为限定本发明的设计思想。在本发明的技术领域里持有相同知识者可以将本发明的技术性思想以多样的形态改良变更,这样的改良及变更应理解为属于本发明的保护范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。