1.本发明涉及病原体鉴定技术领域,尤其涉及一种呼吸机相关性肺炎下呼吸道抽吸物的样本处理方法。
背景技术:
2.呼吸机相关性肺炎指患者接受机械通气48h以后或在拔管48h以内出现的肺炎,是接受机械通气的患者最常见的并发症和死亡原因之一。研究表明,对怀疑患有呼吸机相关性肺炎的患者尽早给予合适的抗菌药物可明显缩短患者患病时间,减少疾病恶化和患者死亡的概率。
3.现有的呼吸机相关性肺炎下呼吸道抽吸物的样本处理通常需要24-48h,若想要获得抽吸物中病原体的药敏结果,则需48-72h,处理时间长,处理效率低。
技术实现要素:
4.鉴于上述的分析,本发明旨在提供一种呼吸机相关性肺炎下呼吸道抽吸物的样本处理方法,用以解决现有呼吸机相关性肺炎患者下呼吸道抽吸物样本处理时间长达24-48h,处理效率低的技术问题。
5.本发明的目的主要是通过以下技术方案实现的:
6.本发明提供了一种呼吸机相关性肺炎下呼吸道抽吸物的样本处理方法,包括以下步骤:
7.s1、样本处理;
8.包括s11.使用皂苷对样本宿主基因进行去除;s12.批量提取dna;
9.s2、构建多样本pcr扩增dna测序文库;
10.s3、对dna测序文库中的dna测序;
11.使用minion纳米孔测序仪进行dna测序,实时进行数据收集;
12.s4、对收集的数据进行筛选和分析;
13.s5、对处理的样本进行病原体基因拼接;
14.s6、对拼接成功的细菌进行耐药性检测。
15.进一步地,在s11中,使用皂苷对样本宿主基因进行去除的过程包括以下子步骤:
16.s111、用250μl无菌pbs缓冲液将临床下呼吸道样本沉淀重悬,充分吹打混匀;
17.s112、加入200μl 5%无菌皂苷溶液使用无菌无酶去离子水溶解,涡旋15s混匀,室温静置10min,以使皂苷溶液和样本充分混合,皂苷可使宿主细胞膨胀破裂;
18.s113、加入350μl无菌无酶去离子水,室温静置30s;
19.s114、加入12μl 5m nacl溶液,上下颠倒混匀,以终止皂苷的膨胀作用;
20.s115、8000rpm 4℃离心5min,弃去上清液;
21.s116、沉淀使用100μl无菌pbs缓冲液重悬,加入100μl hl-san缓冲液,涡旋15s混匀,加入10μl hl-san酶,颠倒混匀,消化游离核酸;
22.s117、37℃水浴,高速震荡15min;
23.s118、8000rpm 4℃离心5min,弃去上清液;
24.s119、加入800μl无菌pbs缓冲液,颠倒混匀,8000rpm 4℃离心5min,弃去上清液;
25.s1110、加入1000μl无菌pbs缓冲液,颠倒混匀,8000rpm 4℃离心5min,弃去上清液。
26.进一步地,在s116步骤中,hl-san缓冲液的配置过程为:将0.2gmgcl
2-6h2o溶于5m nacl中,定容至10ml后使用0.22μm过滤器过滤得到。
27.进一步地,在s12中,使用bscc45s1e试剂盒和自动dna提取仪进行dna提取。
28.进一步地,在s12中,dna提取过程包括以下子步骤:
29.s121、bscc45s1e试剂盒所带溶菌酶全部溶于tet缓冲液中,震荡混匀;
30.s122、180μl混合液重悬沉淀,震荡混匀后37℃孵育30min;
31.s123、向试剂盒第1、7列加入20μl蛋白酶k和含菌重悬液;
32.s124、试剂盒放入机器中,设定程序:s125、待提取完成后使用nanodrop进行dna浓度和纯度鉴定,使用qubit进行浓度精准定量。
33.进一步地,在s2中,使用快速条码试剂盒sqk-rbk004进行多样本快速dna测序文库构建,所有操作均于超净台中进行,所有仪器紫外照射至少30min;所有试剂置于冰上融化,使用前离心处理,吹打混匀。
34.进一步地,在s3中,dna测序过程中,使用芯片引发试剂盒exp-flp002进行芯片引发,所有操作于超净台中进行,所用除芯片外的其他仪器紫外照射30min;使用flo-min106d r9.4芯片和minion测序仪进行测序。
35.进一步地,在s3中,dna测序过程具体包括以下子步骤:
36.s31、将flo-min106d r9.4芯片插入minion测序仪中,连接笔记本电脑,使用minknow软件flowcell test模块进行芯片检测,新芯片纳米孔道数大于800为合格芯片;
37.s32、断开测序仪与电脑的连接,测序仪拿入超净台中上样;
38.s33、向一支试剂flb中加入30μl试剂flt,吹打混匀;
39.s34、打开引发孔,吸出孔中空气;
40.s35、加入800μl flb flt混合试剂,关闭引发孔,室温静置5min;
41.s36、打开引发孔和上样孔,向引发孔中加入200μl flb flt混合试剂,向上样孔中悬空滴加75μl文库;
42.s37、测序仪连接笔记本电脑,使用minknow软件进行测序。
43.进一步地,在s37中,根据使用试剂盒进行程序设定,实时进行数据分析,待出现临床致病菌序列后继续测序1-2h,若无其他致病菌序列出现,则停止测序,断开电脑与测序仪的连接。
44.进一步地,s4步骤中的对收集的数据进行筛选和分析包括:对收集的数据进行筛选和分析的过程包括:先进行碱基判读,碱基判读后进行数据过滤,数据过滤后进行数据质控,并进行宿主基因对比去除,然后进行分类分析。
45.与现有技术相比,本发明至少可实现如下有益效果之一:
46.(1)本发明可针对呼吸机相关性肺炎患者下呼吸道样本进行快速病原体鉴定,将病原体鉴定用时从常规24~48h缩短到6h以内。
47.(2)本发明可有效去除宿主基因组信息,增加纳米孔宏基因组学测序病原体序列获得量。
48.(3)现有细菌耐药性检测需要48-72小时出结果,采用本发明的处理方法只需要6小时就可以获得初步的耐药基因信息,提供耐药和潜在耐药可能的选择。
49.本发明中,上述各技术方案之间还可以相互组合,以实现更多的优选组合方案。本发明的其他特征和优点将在随后的说明书中阐述,并且,部分优点可从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过说明书实施例以及附图中所特别指出的内容中来实现和获得。
附图说明
50.附图仅用于示出具体实施例的目的,而并不认为是对本发明的限制,在整个附图中,相同的参考符号表示相同的部件。
51.图1为本发明的皂苷去除宿主基因的方法与空白对照的对比流程图;图2为本发明的测序和数据分析流程图;图3为本发明的总分析流程和耗时示意图;图4为原始测序数据过滤效果图;其中,a:测序总序列数;b:测序总碱基数;c:最大序列长度;d:最小序列长度;e:平均序列长度。raw data:原始测序数据;filtered data:过滤后数据。
52.图5为minimap2比对剔除宿主基因序列信息效果图;其中,filtered data表示过滤后的数据;without homo genome表示minimap2比对剔除宿主基因序列后的数据。
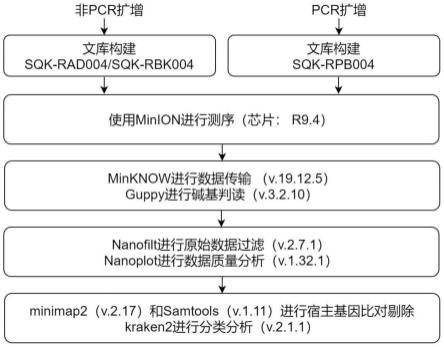
53.图6为本发明的测序数据处理流程示意图;
具体实施方式
54.下面结合附图来具体描述本发明的优选实施例,其中,附图构成本发明的一部分,并与本发明的实施例一起用于阐释本发明的原理,并非用于限定本发明的范围。
55.本发明提供了一种呼吸机相关性肺炎下呼吸道抽吸物的样本处理方法,如图1-3和图6所示,包括以下步骤:
56.s1、样本处理;
57.包括以下子步骤:s11.使用皂苷对样本宿主基因进行去除;s12.批量提取dna;
58.s2、利用提取的dna进行建库,得到dna测序文库;
59.使用sqk-rbk004试剂盒进行dna建库;
60.s3、对dna测序文库中的dna进行测序;
61.使用minion纳米孔测序仪进行测序,实时进行数据收集;
62.s4、对收集的数据进行筛选和分析;
63.s5、对处理样本进行病原体基因拼接;
64.s6、对拼接成功的细菌进行耐药性检测。
65.在s1步骤中,在使用皂苷对样本宿主基因去除之前,先进行样本的收集和保存,具体过程为:临床收集呼吸机相关性肺炎患者的下呼吸道抽吸物,每个患者至少收集固形物1ml,收集好后立刻进行后续处理,若不能立刻处理,则样本在4℃最多保存4小时。需要注意的是,首先,采样后冰上运输,加入4倍体积无菌pbs缓冲液;其次,抽吸物样本放入37℃水浴中,高速震荡至少30min,直至痰痂完全溶解;再次,将溶解后的样本分装为1ml一支,装入
1.5ml无菌ep管中,8000rpm4℃离心30min;最后,将上清液收集于5ml冻存管中,样本液氮急冻2-3min后-80℃保存。
66.需要说明的是,微生物培养过程包括:
67.s1’、将-80℃保存样本室温复温;
68.s2’、挑取5μl样本,按照三区法在増菌培养基上划线,37℃培养过夜培养12-16小时;
69.s3’直接使用平板长出的单菌落进行质谱分析细菌种类鉴定和微生物耐药表型鉴定;
70.s4’挑取1μl单菌落样本,溶于増菌肉汤培养基中,37℃水浴中速震荡培养6-8小时;
71.s5’含菌肉汤吹打混匀,1ml每支分装于1.5ml无菌ep管中,13000rpm 4℃离心1分钟;
72.s6’上清收集到感染物专用废液缸中,收集纯菌,液氮急冻2-3分钟,-80℃保存,用作后续基因组学研究。
73.在s11步骤中,抽吸物样本收集后,使用皂苷对样本宿主基因进行去除,具体包括以下子步骤:
74.s111、用250μl无菌pbs缓冲液将临床下呼吸道样本沉淀重悬,充分吹打混匀;
75.s112、加入200μl 5%无菌皂苷溶液使用无菌无酶去离子水溶解,涡旋15s混匀,室温静置10min,以使皂苷溶液和样本充分混合,皂苷可使宿主细胞膨胀破裂;
76.s113、加入350μl无菌无酶去离子水,室温静置30s;
77.s114、加入12μl 5m nacl溶液,上下颠倒混匀,以终止皂苷的膨胀作用;
78.s115、8000rpm 4℃离心5min,弃去上清液;
79.s116、沉淀使用100μl无菌pbs缓冲液重悬,加入100μl hl-san缓冲液,涡旋15s混匀,加入10μl hl-san酶,颠倒混匀,消化游离核酸;
80.s117、37℃水浴,高速震荡15min;
81.s118、8000rpm 4℃离心5min,弃去上清液;
82.s119、加入800μl无菌pbs缓冲液,颠倒混匀,8000rpm 4℃离心5min,弃去上清液;
83.s1110、加入1000μl无菌pbs缓冲液,颠倒混匀,8000rpm 4℃离心5min,弃去上清液。
84.在上述s116步骤中,hl-san缓冲液的配置过程为:将0.2gmgcl
2-6h2o溶于5m nacl中,定容至10ml后使用0.22μm过滤器过滤得到。
85.需要说明的是,本发明的皂苷去宿主过程的原理为:皂苷作为一种洗涤剂,可通过渗透作用使无细胞壁细胞的细胞膜膨胀破裂,而不会对有细胞壁的细菌产生影响。无壁细胞的细胞膜破裂以后,游离的dna和rna可被dna水解酶和rna水解酶降解,从而降低宿主基因的丰度。
86.在s11步骤中,当使用皂苷对样本宿主基因进行去除时,同时进行了空白对照试验,如图1所示,空白对照试验包括以下过程:
87.s111’、用250μl无菌pbs缓冲液将临床下呼吸道样本(eta样本)沉淀重悬,充分吹打混匀;
88.s112’、加入200μl无菌无酶去离子水溶解,室温静置10min;
89.s113’、加入350μl无菌无酶去离子水,室温静置30s;
90.s114’、加入12μl无菌无霉水;
91.s115’、8000rpm 4℃离心5min,弃去上清液;
92.s116’、沉淀使用100μl无菌pbs缓冲液重悬,加入110μl无菌pbs缓冲液;
93.s117’、37℃水浴,高速震荡15min;
94.s118’、8000rpm 4℃离心5min,弃去上清液;
95.s119’、加入800μl无菌pbs缓冲液,颠倒混匀,8000rpm 4℃离心5min,弃去上清液;
96.s1110’、加入1000μl无菌pbs缓冲液,颠倒混匀,8000rpm 4℃离心5min,弃去上清液。
97.对照组:对比皂苷去宿主基因效果,皂苷去宿主基因组使用标准流程,对照组将所有试剂更换为无菌无酶水。其中,eta:下呼吸道抽吸物;hl-san:热敏性盐活性核酸酶。
98.在s12步骤中,使用bscc45s1e试剂盒(博日公司生产)和自动dna提取仪(博日公司生产)进行dna提取。
99.上述s12步骤包括以下子步骤:
100.s121、bscc45s1e试剂盒所带溶菌酶全部溶于tet缓冲液中,震荡混匀;其中,tet缓冲液为bscc45s1e试剂盒附带的试剂。
101.s122、180μl混合液重悬沉淀,震荡混匀后37℃孵育30min;
102.s123、向bscc45s1e试剂盒第1列和第7列加入20μl蛋白酶k和含菌重悬液;其他列用于盛放自动dna提取的其它试剂,例如,自动dna提取仪会自动筛选所需试剂;
103.s124、试剂盒放入自动dna提取仪中,设定程序,如表3所示:
104.表1全自动dna提取装置设置时间表
[0105][0106]
s125、待提取完成后使用nanodrop进行dna浓度和纯度鉴定,获得满足minion纳米孔测序要求质量的dna溶液,使用qubit进行浓度精准定量。
[0107]
在s2步骤中,使用快速条码试剂盒sqk-rbk004进行多样本快速pcr扩增dna测序文
库构建,所有操作均于超净台中进行,所用仪器紫外照射30min;所有试剂置于冰上融化,使用前瞬时离心,吹打混匀。
[0108]
利用qubit dna浓度测定:使用dsdna hs assay kit for试剂盒(12640es60,翌圣生物科技股份有限公司)进行dna浓度测定,测定步骤为:
[0109]
1.准备0.5ml无菌无酶ep管,管盖上标注标准品1、2和样品编号;
[0110]
2.按照qubit reagent:qubit hs dna buffer=1:200的比例配制缓冲液,按照每个样品200μl的总量进行配制,涡旋混匀;
[0111]
3.标准品管加入190μl缓冲液和10μl标准品,样品管中加入199μl缓冲液和1μl样品,涡旋混匀;
[0112]
4.室温静置2分钟;
[0113]
5.qubit开机,选择“dna”,“1hs dna high sensitivity”,“read standards”;
[0114]
6.放入标准品1,点击read进行读数;
[0115]
7.放入标准品2,点击read进行读数;
[0116]
8.选择“run samples”,选择加入1μl样品,输出单位为ng/μl;
[0117]
9.放入样品管,点击“read tube”;
[0118]
10.记录样本浓度。
[0119]
在上述s2步骤中,多样本pcr扩增dna测序文库构建;具体包括以下过程:
[0120]
s21、分别向无菌无酶0.2ml pcr管中加入5ng dna,dna体积不超过3μl;
[0121]
s22、加入无菌无酶去离子水,补齐至3μl;
[0122]
s23、分别加入1μl试剂frm,轻弹混匀,瞬时离心;
[0123]
s24、30℃孵育1分钟;
[0124]
s25、80℃孵育1分钟,冰上降温;
[0125]
s26、加入23.5μl无菌无酶去离子水,1μl pcr条码试剂rlb01-12a,10μl 5x q5缓冲液,1μl dntp,0.5μl q5 polymerase,10μl增强子;
[0126]
s27、按照表3.4所述程序进行扩增;
[0127]
表2多样本pcr扩增文库构建扩增设置时间表
[0128][0129]
s28、ampure xp磁珠涡旋混匀,所有样本收集于一个无菌无酶1.5ml ep管中,按照
每个样本30μl的体积加入混匀后的磁珠;
[0130]
s29、水平摇床室温200转摇5分钟;
[0131]
s210、ep管放入磁力架上,待磁珠完全吸附,液体澄清后,轻柔吸去上清液;
[0132]
s211、加入200μl 75%乙醇清洗磁珠,弃去洗液;
[0133]
s212、重复步骤11;
[0134]
s213、从磁力架上取下ep管,瞬时离心,吸去剩余乙醇液体;
[0135]
s214、开盖静置2-5分钟,干燥磁珠;
[0136]
s215、加入10μl te缓冲液,充分吹打重悬磁珠,室温静置2分钟;
[0137]
s216、ep管放入磁力架上,收集纯化dna于无菌无酶0.2ml pcr管中;
[0138]
s217、加入1μl试剂rap,轻弹混匀,瞬时离心,室温静置5分钟;
[0139]
s218、加入34μl试剂sqb,25.5μl试剂lb,4.5μl无菌无酶去离子水,轻弹混匀,瞬时离心。
[0140]
需要说明的是,在上述s3步骤中,dna测序过程具体包括以下子步骤:
[0141]
s31、将flo-min106d r9.4芯片插入minion测序仪中,连接笔记本电脑,使用minknow软件flowcell test模块进行芯片检测,新芯片纳米孔道数大于800为合格芯片;
[0142]
s32、断开测序仪与电脑的连接,测序仪拿入超净台中上样;
[0143]
s33、向一支试剂flb中加入30μl试剂flt,吹打混匀,得到flb flt混合试剂;flb和ftl均为bscc45s1e试剂盒附带的试剂;
[0144]
s34、打开引发孔,吸出孔中空气;
[0145]
s35、加入800μl上述的flb flt混合试剂,关闭引发孔,室温静置5min;
[0146]
s36、打开引发孔和上样孔,向引发孔中加入200μl flb flt混合试剂,向上样孔中悬空滴加75μl文库;
[0147]
s37、测序仪连接笔记本电脑,使用minknow软件进行测序。
[0148]
在s37中,根据使用试剂盒进行程序设定,实时进行数据分析,待出现临床致病菌序列后继续测序1-2h,若无其他致病菌序列出现,则停止测序,断开电脑与测序仪的连接。
[0149]
在上述步骤3中,使用芯片清洗试剂盒(exp-wsh004,英国牛津纳米孔科技有限公司)进行芯片清洗,所有操作于超净台中进行,所用除芯片外的其他仪器紫外照射30分钟,具体清洗过程包括:第一、采用2μl试剂wmx和398μl试剂dil混合成清洗混合液,吹打混匀;第二、打开引发孔,使用1000μl移液枪调至800μl量程,对准引发孔插入后逆旋20-30μl,至储存液进入吸头底部,吸出孔中空气;第三、向引发孔中加入400μl清洗混合液,关闭引发孔,室温静置30分钟;第四、打开引发孔,向引发孔中加入500μl储存试剂s,关闭引发孔;第五、从废液孔中吸净废液;第六、芯片放回包装袋中,4℃保存。
[0150]
在上述s4步骤中,对收集的数据进行筛选和分析的过程包括:先进行碱基判读,碱基判读后进行数据过滤,数据过滤后进行数据质控,并进行宿主基因对比去除,然后进行分类分析。
[0151]
进一步地,使用guppy进行碱基判读,使用nanofilt进行数据过滤,使用nanoplot进行数据质控,使用minimap2进行宿主基因比对去除,使用kraken2进行分类分析,所有分析都可在本地服务器上进行,4-6min左右即可完成所有分析,减少了数据上传英国伦敦服务器导致的潜在数据泄露风险。
[0152]
在上述s4步骤中,利用guppy进行碱基判读,碱基判读和数据质控的实现过程为:编写脚本guppy.sh,对minion测序下机的原始数据文件进行碱基判读。碱基判读使用guppy(v.3.2.10)进行,原始数据质量统计使用美国密西根大学dickson实验室开发的summarizefastq.pl程序进行,原始数据质量可视化分析(数据质控)使用nanoplot(v.1.32.1)进行。需要说明的是,碱基判读的具体实施过程如实施例1所示。
[0153]
上述s4步骤中的数据过滤过程包括:编写脚本datafilt.sh,对原始数据中质量值(q值)小于7的低质量序列进行剔除,并切掉接头序列(头部接头序列150bp,尾部接头序列50bp)。数据过滤使用nanofilt(v.2.7.1)进行,过滤后数据质量统计使用美国密西根大学dickson实验室开发的summarizefastq.pl程序进行,数据质量可视化分析使用nanoplot(v.1.32.1)进行。需要说明的是,数据过滤及数据质控的实施过程参见实施例2。
[0154]
数据过滤结果:对于测序获得的原始数据进行数据过滤,得到可用于测序的数据。过滤后所有样本的序列数和总碱基数均有所降低,绝大多数序列最大长度减少200bp,序列最小长度由100bp左右增加至500bp,平均序列长度有程度不一的升高,参见图4。
[0155]
在上述s4步骤中,宿主基因比对及剔除过程包括:
[0156]
编写脚本mapping.sh,对数据过滤及数据质控后的测序数据进行宿主基因比对及剔除。数据比对使用minimap2(v.2.17)进行,数据筛选使用samtools(v.1.11)。比对基因组为ncbi下载的为grch38人类基因组序列。需要说明的是,宿主基因对比去除的具体过程参见实施例3。
[0157]
宿主基因数据剔除结果为:对测序数据使用minimap2比对的方法再次进行宿主基因剔除,获得丰度较高的病原体序列信息。如图5所示,所有样本宿主基因信息都有不同程度的减少,20例样本中所有的宿主基因信息都被剔除,仅余微生物基因序列信息,为后续分类比对和物种鉴定提供了更精准的数据,减少了分类比对用时。
[0158]
在上述s4步骤中,利用kraken2进行分类比对和分析,其过程为:编写脚本kraken2_classification.sh,使用kraken2(v.2.1.1)程序对病原体序列信息进行分类比较。
[0159]
上述s5步骤中,对处理样本进行病原体基因拼接,其过程为:
[0160]
米孔测序数据病原体基因组拼接:
[0161]
编写脚本bacassemble_ont.sh,对样本nanopore测序数据结果进行目的病原体序列筛选和基因组拼接。拼接使用flye(2.8.3)程序进行,校正纠错使用medaka(v.1.4.2)进行。
[0162]
上述s6步骤中,对拼接成功的细菌进行耐药性检测,包括:对基因拼接成功的细菌使用网页版resistance gene identifier(https://card.mcmaster.ca/analyze/rgi)进行病原体耐药基因鉴定。
[0163]
1.在select data type处选择dna sequence;
[0164]
2.在upload fasta sequence file(s)处上传拼接好的文件;
[0165]
3.在select criteria处选择perfect and strict hits only;
[0166]
4.在nudge≥95%identity loose hits to strict处选择exclude nudge;
[0167]
5.在sequence quality处选择high quality/coverage;
[0168]
6.点击submit,获得分析结果,结果判定为perfect和strict,匹配区域一致性百
分比(%identity of matching region)大于90%,同时占参考基因长度百分比(%length of reference sequence)大于80%的耐药基因被认为是合格耐药基因。
[0169]
为了对测序结果进行验证,以观察其准确性,包括以下过程:
[0170]
(1)利用qrt-pcr对测序结果进行验证。
[0171]
使用翌圣公司的hieffuniversal blue qpcr sybr master mix(11184es08)进行qrt-pcr,操作全程避光,使用无菌无酶未高压的吸头和ep管进行操作,操作方法按照说明书所述进行:
[0172]
1.委托睿博兴科公司进行引物合成,引物序列见表3;
[0173]
2.按照每个样本每个目的基因3个复孔的体积进行混合液配制,每个孔混合液含量如下:10μl sybr 0.4μl正引物 0.4μl反引物 7.2μl无菌无酶去离子水;
[0174]
3.按照每个孔18μl混合液 2μl dna的量进行加样,每次加样设置1个阴性对照组,1个空白对照组和1个阳性对照组;
[0175]
表3引物序列
[0176][0177][0178]
(2)进行扩增时间,以对测序结果进行验证。
[0179]
按表4所述方法设定pcr扩增程序;
[0180]
表4 pcr扩增程序时间设置表
[0181][0182]
样本结果ct值大于35或阴性对照组认定为样本阴性,ct值介于30~35或30~阴性对照组之间认定为可疑,ct值小于30认定为样本阳性。
[0183]
需要说明的是,本发明中的样本中病原体诊断标准如下:
[0184]
1.微生物培养:三区法划线,37℃培养24-48小时后出现肉眼可见菌落定义为阳性。细菌培养半定量结果以三区肉眼可见菌落数进行区分;
[0185]
表5三区法细菌半定量结果定义
[0186][0187]
2.qrt-pcr:
[0188]
1)阳性:ct值<30,同时小于阴性对照组,结果具有统计学意义;
[0189]
2)阳性可疑:ct值处于30~35之间,小于阴性对照组,结果具有统计学意义;
[0190]
3)阴性:ct值高于35,或高于阴性对照组或与阴性对照组无统计学差异。
[0191]
3.宏基因组学:
[0192]
1)阳性:宏基因组学结果显示病原体序列数大于1条,且同时大于所有病原体序列数的1%;
[0193]
2)阳性可疑:病原体序列数只有1条,但大于所有病原体序列数的10%
[0194]
4.样本中病原体存在判断标准:
[0195]
1)微生物培养阳性;
[0196]
2)qrt-pcr阳性;
[0197]
3)qrt-pcr阳性可疑合并宏基因组学阳性或阳性可疑。
[0198]
实施例1
[0199]
碱基判读实现过程中,其具体脚本代码为:
[0200]
#!/bin/bash
[0201]
module load conda/miniconda2
[0202]
read-p"enter sample id:"sampleid#输入样本id
[0203]
#使用guppy进行碱基判读
[0204]
time guppy_basecaller\
[0205]
‑‑
input_path#input_path\#设置数据输入路径,即原始fast5数据保存路径
[0206]
‑‑
recursive\#递归处理
[0207]
‑‑
save_path#output_path\#设置输出数据保存路径,即判读fastq数据保存路径
[0208]
‑‑
flowcell flo-min106\#测序芯片编号
[0209]
‑‑
kit#kitid_for_library_built\#建库试剂盒货号
[0210]
‑‑
device'cuda:0'#测序设备;
[0211]
cat*.fastq》》${sampleid}.total.fastq#将样本所有数据整合为一个fastq文件;
[0212]
summarizefastq.pl${sampleid}.total.fastq》
[0213]
${sampleid}summarize_rawdata.txt
[0214]
#使用美国密西根大学dickson实验室开发的summarizefastq.pl脚本进行原始数据统计
[0215]
统计数据保存于${sampleid}summarize_rawdata.txt文件。
[0216]
实施例2
[0217]
本实施例提供了一种数据过滤及数据质控的具体实施过程,具体脚本代码为:
[0218]
#!/bin/bash
[0219]
module load conda/miniconda2
[0220]
read-p"enter sample id:"sampleid#输入样本id
[0221]
source activate nanopack#激活数据分析环境;
[0222]
time nanofilt\#使用nanofilt进行数据过滤并计算用时
[0223]
${sampleid}.total.fastq\#输入待过滤数据
[0224]-l 500\#过滤掉小于500bp的序列
[0225]-q 7\#最低平均质量值
[0226]
过滤掉质量值小于7的低质量序列
[0227]
‑‑
headcrop 150\#从头部切掉150bp接头序列
[0228]
‑‑
tailcrop 50\#从尾部切掉50bp结尾序列
[0229]
》${sampleid}.clean.fastq\#结果输出至${sampleid}.clean.fastq文件;
[0230]
summarizefastq.pl${sampleid}.clean.fastq》
[0231]
${sampleid}summarize_cleandata.txt
[0232]
time nanoplot\#使用nanoplot进行数据质量可视化分析
[0233]
‑‑
threads 6\#分析使用线程数
[0234]
‑‑
outdir nanoplotsummary\#输出结果目录
[0235]
‑‑
n50\#在序列读长直方图中显示n50的标识
[0236]
‑‑
title${sampleid}.clean\#设置输出图形的标题为${sampleid}.clean
[0237]
‑‑
fastq${sampleid}.clean.fastq\#待分析的文件为
[0238]
${sampleid}.clean.fastq
[0239]
‑‑
plots hex dot kde pauvre\#文件绘图类型:kde(等高线图)、dot(点图)、hex(颜色深浅六角形图)、pauvre(双侧有坐标轴的柱状图)、
‑‑
color green#点和柱状图的颜色为绿色;、conda deactivate#退出数据分析环境。
[0240]
实施例3
[0241]
本实施例提供了一种宿主基因对比及剔除的具体实施过程。其具体脚本代码为:
[0242]
#!/bin/bash
[0243]
module load conda/miniconda2
[0244]
read-p"enter sample id:"sampleid#输入样本id
[0245]
source activate minimap2
[0246]
#下载并构建人源基因组索引
[0247]
一次构建成功后无需重复构建。
[0248]
wget
[0249]
https://ftp.ncbi.nlm.nih.gov/refseq/h_sapiens/annotation/grch38_latest/refseq_identifiers/grch38_latest_genomic.fna.gz~/minimap2/#下载
[0250]
grch38人源基因组序列到~/minimap2/路径;
[0251]
minimap2-d~/minimap2/human.min
[0252]
~/minimap2/grch38_latest_genomic.fna.gz#以
[0253]
grch38_latest_genomic.fna.gz为参考基因组
[0254]
使用minimap2构建人源基因组索引human.min;
[0255]
#使用minimap2进行测序数据比对分析。
[0256]
minimap2-ax map-ont\#设置测序仪器为ont
[0257]
输出文件格式为sam
[0258]
~/minimap2/human.min\#设置参考索引
[0259]
${sampleid}.clean.fastq\#设置输入文件
[0260]
》${sampleid}.clean.sam\#设置输出文件;
[0261]
samtools view\#使用samtools view进行文件格式转换
[0262]-sb\#输入sam格式文件
[0263]
输出bam格式文件
[0264]-o${sampleid}.clean.bam\#设置输出文件
[0265]
${sampleid}.clean.sam\#设置输入文件;
[0266]
#使用samtools进行比对序列处理及病原体序列提取。
[0267]
samtools sort\#使用samtools sort对上述文件进行排序
[0268]-o bam\#输出文件格式
[0269]-o${sampleid}.clean.sorted.bam\#设置输出文件
[0270]
${sampleid}.clean.bam\#设置输入文件;
[0271]
samtools view-f 4${sampleid}.clean.sorted.bam》
[0272]
${sampleid}.nohuman.bam#使用samtools view提取未比对上的非人源基因组序列;
[0273]
samtools fastq${sampleid}.nohuman.bam》
[0274]
${sampleid}.nohuman.fastq#使用samtools fastq将
[0275]
${sampleid}.nohuman.bam文件转化为${sampleid}.nohuman.fastq文件,获得无人源基因组的病原体序列信息;
[0276]
conda deactivate#退出分类分析环境。
[0277]
实施例4
[0278]
本实施例提供了对经过宿主基因对比去除后的病原体序信息分类分析的具体实施过程。其具体脚本代码为:
[0279]
#!/bin/bash
[0280]
module load conda/miniconda2
[0281]
read-p"enter sample id:"sampleid#输入样本id
[0282]
source activate classification
[0283]
#构建kraken2标准参考基因组数据库
[0284]
一次构建好无需重复构建
[0285]
但需不定时更新数据库。
[0286]
mkdir standardk2idx#建立索引路径;
[0287]
kraken2-build
‑‑
standard
‑‑
threads 24
‑‑
db standardk2idx#从ncbi上下载所有构建相关taxonomy等文件并构建索引
[0288]
选择使用24个线程来进行处理;
[0289]
#使用kraken2进行病原体分类信息分析
[0290]
recordstats kraken2\#使用kraken2进行分类分析并记录分析过程服务器状态和耗时
[0291]
‑‑
db standardk2idx\#使用标准参考基因组数据库进行分类比对
[0292]
‑‑
threads 10\#使用10个线程来进行处理
[0293]
‑‑
output${sampleid}.cleanout.txt\#序列分析结果输出文件为
[0294]
${sampleid}.cleanout.txt
[0295]
‑‑
report${sampleid}.cleanreport.txt\#分类分析结果输出文件为
[0296]
${sampleid}.cleanreport.txt
[0297]
${sampleid}.clean.fasta#待分析文件为${sampleid}.clean.fasta;
[0298]
grep-w s${sampleid}report.txt|sort-n-k 3》
[0299]
${sampleid}_species.txt#抓取结果中的种信息并将其按照reads数的多少进行排序
[0300]
结果输出至${sampleid}_species.txt文件中;
[0301]
conda deactivate#退出分类分析环境。
[0302]
实施例5
[0303]
本实施例提供了一种基因拼接过程,基因拼接的具体脚本代码为:
[0304]
#!/bin/bash
[0305]
module load conda/miniconda2
[0306]
read-p"enter sample id:"sampleid#输入样本id
[0307]
read-p"enter abbreviation of pathogene:"abbre#输入待检测病原体的缩写
[0308]
read-p"enter abbreviation of pathogene:"taxonyid#输入待检测病原体的物种分类号
[0309]
#用于病原体fastq序列筛选
[0310]
从kraken2分析结果中挑出病原体的fastq序列
[0311]
#从所有比对分析结果中找出病原体序列整合成新的文件
[0312]
mkdir${abbre}
[0313]
cd kraken2/
[0314]
awk'{if($3=='${taxonyid}')printf$2"\n"}'
[0315]
${sampleid}_clean_nohumanout.txt》${sampleid}${abbre}titleout.txt
[0316]
line=$(cat${sampleid}${abbre}titleout.txt|wc-l)
[0317]
cd..
[0318]
for((i=1;i《=$
131
;i ))#for循环语句
[0319]
do
[0320]
title=$(sed-n${i}p kraken2/${sampleid}${abbre}titleout.txt)#抓取筛选出的目的病原体title文件中的第n行
[0321]
grep"${title}"${sampleid}_clean_nohuman.fastq-a 3》》
[0322]
${abbre}/${sampleid}${abbre}.fastq#使用title进行序列抓取并抓取标题后的三行(总共4行)至指定文件中
[0323]
done
[0324]
#将fastq文件转换为fasta文件
[0325]
用以下一步操作
[0326]
cd${abbre}
[0327]
source activate nanopack
[0328]
seqtk seq-a${sampleid}${abbre}.fastq》${sampleid}${abbre}.fasta#fastq文件转换为fasta文件
[0329]
conda deactivate
[0330]
#用flye进行基因组拼接
[0331]
source activate minimap2
[0332]
mkdir antibiotic
[0333]
mkdir antibiotic/flye
[0334]
flye
‑‑
nano-raw${sampleid}${abbre}.fasta-o antibiotic/flye
[0335]
#用medaka进行组装结果纠错
[0336]
cd antibiotic
[0337]
mkdir medaka
[0338]
medaka_consensus-i../${sampleid}${abbre}.fasta-d flye/assembly.fasta-o
[0339]
medaka/-t 4》medaka/medaka.log
[0340]
#-i输入拼接前的原始fasta文件
[0341]
#-d输入拼接后的fasta文件
[0342]
#-o组装结果纠错结果输出目录
[0343]
#-t使用的threads数
[0344]
conda deactivate。
[0345]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。