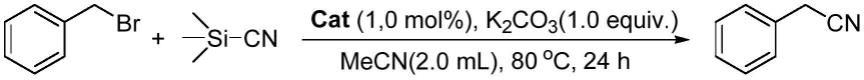
1.本发明涉及催化合成技术领域,具体涉及一种苯乙腈的多酸催化制备方法。
背景技术:
2.氰基是有机化学的重要结构单元,广泛存在于医药、农药、染料、光电材料中,是其有效活性官能团,在其中起着不可替代的作用。而苯乙腈是重要的医药、农药及化工中间体,主要用于合成青霉素、苯巴比妥、地巴唑、吲哚波芬、苯基异丙胺等医药产品;生产苯乙酸、苯乙胺、二苯乙腈、苯乙醇、苯乙醛、邻乙苯基苯甲酸等有机化学品;用于制造辛硫磷、稻丰散等农药品;还用于生产染料等等。
3.目前主要有两种合成方法:一是溶剂法合成:氯化苄和氰化钠在乙醇中进行亲核取代反应,以氯化苄和液体氰化钠为原料,在相转移催化剂作用下采用间歇生产方式,在反应釜内,在温度90-100℃条件下,向氯化苄中缓慢加入30%氰化钠溶液,加料结束后继续保温l-3h,分液、精馏得到产品苯乙腈。工艺中氰化过程存在收率低(85-90%)、处理量小、副反应较多等问题,除此之外使用固体氰化钠成本较高,需对溶剂进行回收,因而工艺路线时间长。而当氯化苄纯度较差时,收率大幅度下降。二是水法合成:以水代替乙醇,以二甲胺作催化剂,该法存在收率低,质量不高,苯乙腈易变色等缺点。另外氰化产物在特定的条件下还可以转化成含有氨基、酰胺基、羧基、醛基、醛肟和四氮唑基等结构单元的有机化合物。
4.苯乙腈衍生物广泛用于医药、农药等功能化合物的制备。盐酸文拉法辛是由对甲氧基苯乙腈经一系列反应合成,具有抗抑郁的作用;阿那曲唑中含有苯乙腈结构单元,具有抗肿瘤的作用;盐酸异波帕明是由3,4-二甲氧基苯乙腈为原料合成,具有治疗心脑血管疾病的功效;酮洛芬也可以由苯乙腈合成,其具有抗炎镇痛的功效。另外,苯乙腈衍生物在染料、有机光电材料方面也都有着很好的应用。
5.由于人们环保意识的不断加强,苯乙腈在欧美生产已受到严格限制,高纯度的苯乙腈需求量越来越大,低含量的工艺将逐步被淘汰,这也意味着我们开发出一种高纯度苯乙腈的合成路线迫在眉睫。
技术实现要素:
6.本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种高活性、高选择性、反应条件温和、绿色、环保,催化剂可以回收利用,具有极大工业化生产潜力的苯乙腈的多酸催化制备方法。
7.本发明的目的可以通过以下技术方案来实现:
8.发明人了解到,多金属氧酸盐(多酸)作为一种新型的高效的多功能催化剂,即有酸催化性能,又具有氧化还原催化性能,且稳定性很好,既可作均相反应,也可作非均相反应,甚至可作相转移催化剂,是一类很有前景的绿色、环保催化剂,广泛应用于催化、分析、药物、电化学、光化学和石油化学等领域。具有高活性、高选择性、高稳定性、可循环利用的优势,提出以下方案:
9.本发明以溴化苄为原料,多金属氧酸盐(lindquist型和anderson型,其中以anderson型骨架的fe、al、cr、ni、mn、cu和co等中心金属为主)为催化剂。该催化体系可以将溴化苄和三甲基氰硅烷催化氧化为苯乙腈,具有高活性和高选择性,反应条件温和、绿色和环保,催化剂可以回收利用,具有极大的工业化生产潜力,具体方案如下:
10.一种苯乙腈的多酸催化制备方法,该方法包括以下步骤:
11.将催化剂多金属氧酸盐和添加剂放进洁净干燥的反应器中;
12.将溴化苄和三甲基氰硅烷依次加入反应器中;
13.加入溶剂,加热搅拌反应后,分离得到苯乙腈,反应式如下:
14.进一步地,所述的多金属氧酸盐包括lindquist型或anderson型的多金属氧酸盐,所述催化剂的中心金属包括fe、al、cr、ni、mn、cu或co。
15.进一步地,所述的添加剂包括kcl、k2co3、khco3、na2co3、nahco3或nabr中的一种或多种,优选k2co3。
16.进一步地,所述的溶剂包括乙腈、丙酮、二丁醚、dmf、苯、1,4-二氧六环或dmso中的一种或多种,优选乙腈。
17.进一步地,所述的溴化苄和三甲基氰硅烷摩尔比为1:(1.0-2.0),优选1:2。
18.进一步地,所述催化剂的添加量为溴化苄的0.5-1.5mol%,优选1.0mol%。
19.进一步地,所述添加剂的添加量为反应量的1.0-4.0当量,即添加剂与溴化苄的摩尔比为(1.0-4.0):1,优选1.0当量。
20.进一步地,所述的溶剂与溴化苄的比为(2.0-6.0)ml:1mmol。
21.进一步地,所述加热的温度为50-80℃,优选80℃,反应的时间为12-24h,优选24h。
22.进一步地,该方法还包括以下步骤:将反应结束后的体系进行萃取过滤,取下层水相,将水相处理后进行回收,将回收的多金属氧酸盐再用于溴化苄和三甲基氰硅烷制备苯乙腈反应中。
23.与现有技术相比,本发明具有以下优点:
24.(1)本发明反应物转化率高、产物收率高、无三废、生产成本低等特点,是一种原子经济性高、环境友好型的制备苯乙腈的方法;
25.(2)本发明所用的催化剂是一种新型催化剂——多金属氧酸盐(杂多酸),经过简单处理后还可以循环使用多次;
26.(3)本发明反应过程中不需要额外添加酸,对仪器损害小,降低了工业生产的设备折旧费用,十分有利于工业生产,具有潜在的应用前景。
附图说明
27.图1为实施例1中苯乙腈的核磁共振碳谱1h nmr(cdcl3);
28.图2为实施例1中苯乙腈的核磁共振氢谱
13
c nmr(cdcl3)。
具体实施方式
29.下面结合附图和具体实施例对本发明进行详细说明。本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
30.一种苯乙腈的多酸催化制备方法,该方法包括以下步骤:
31.将催化剂多金属氧酸盐和添加剂放进洁净干燥的反应器中;多金属氧酸盐包括lindquist型或anderson型的多金属氧酸盐,所述催化剂的中心金属包括fe、al、cr、ni、mn、cu或co;添加剂包括kcl、k2co3、khco3、na2co3、nahco3或nabr中的一种或多种,优选k2co3。添加剂的添加量为反应量的1.0-4.0当量,即添加剂与溴化苄的摩尔比为(1.0-4.0):1,优选1.0当量。
32.将溴化苄和三甲基氰硅烷依次加入反应器中;溴化苄和三甲基氰硅烷摩尔比为1:(1.0-2.0),优选1:2;催化剂的添加量为溴化苄的0.5-1.5mol%,优选1.0mol%。
33.加入溶剂,加热搅拌反应后,分离得到苯乙腈。溶剂包括乙腈、丙酮、二丁醚、dmf、苯、1,4-二氧六环或dmso中的一种或多种,优选乙腈。溶剂与溴化苄的比为(2.0-6.0)ml:1mmol;加热的温度为50-80℃,优选80℃,反应的时间为12-24h,优选24h。
34.另外,还可以将反应结束后的体系进行萃取过滤,取下层水相,将水相处理后进行回收,将回收的多金属氧酸盐再用于溴化苄和三甲基氰硅烷制备苯乙腈反应中。
35.为了对本发明进行进一步的详细说明,下面给出几个具体实施案例,案例主要以不同金属原子为中心的anderson型多金属氧酸盐催化剂为例。但是,本发明不限于这些实施例。
36.实施例1
37.将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的cu为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,反应式如下:
[0038][0039]
制样做gc-ms检测,根据gc-ms结果,得知反应产率为98%。分离提纯后进行核磁测试,如图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0040]
实施例2
[0041]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的al为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为80%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0042]
实施例3
[0043]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的cr为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为74%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0044]
实施例4
[0045]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的co为中心金属的
anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为87%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0046]
实施例5
[0047]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的ni为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为64%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0048]
实施例6
[0049]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的mn为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为57%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0050]
实施例7
[0051]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的fe为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为72%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0052]
实施例8
[0053]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的zn为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为86%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0054]
实施例9
[0055]
将0.171g的溴化苄、0.198g的三甲基氰硅烷、1.0mol%的v为中心金属的anderson型多金属氧酸盐、1.0当量的添加剂k2co3、2.0ml的溶剂乙腈投入到干燥反应管中,反应管上套氧气球,反应温度控制在80℃,保温反应24h后,停止反应,冷却到室温,制样做gc-ms检测,根据gc-ms结果,得知反应产率为48%。分离提纯后进行核磁测试,参考图1-2,由所得的氢谱、碳谱数据证明了其为苯乙腈。
[0056]
实施例10
[0057]
反应步骤同实例1,但是不同之处在于,所用催化剂为实施例1回收后的催化剂第1次使用,gc-ms分析得知溴化苄的转化率大于95%,分离提纯得到产物,核磁确认为苯乙腈,收率95%。
[0058]
实施例11
[0059]
反应步骤同实例1,但是不同之处在于,所用催化剂为实施例1回收后第2次使用,gc-ms分析得知溴化苄的转化率大于93%,分离提纯得到产物,核磁确认为苯乙腈,收率92%。
[0060]
实施例12
[0061]
反应步骤同实例1,但是不同之处在于,所用催化剂为实施例1回收后第3次使用,gc-ms分析得知溴化苄的转化率大于92%,分离提纯得到产物,核磁确认为苯乙腈,收率90%。
[0062]
实施例13
[0063]
反应步骤同实例1,但是不同之处在于,所用催化剂为实施例1回收后第4次使用,gc-ms分析得知溴化苄的转化率大于90%,分离提纯得到产物,核磁确认为苯乙腈,收率89%。
[0064]
实施例14
[0065]
反应步骤同实例1,但是不同之处在于,所用催化剂为0.1mol%lindquist型[(c4h9)4n]3[v
10o28
h3],gc-ms分析得知溴化苄的转化率为73%,分离提纯得到产物,经核磁图谱确认为苯乙腈。
[0066]
所有上述的实施方案,并没有设定实施这种新产品或新方法的其他形式。本领域的技术人员将利用这一重要信息,上述内容修改,以实现类似的执行情况。但是,所有给予本发明的修改或改造属于本发明保留的权利。
[0067]
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。