一种近红外比率型ph荧光探针及其制备方法与其在破骨吸收成像中的应用
技术领域
1.本发明属于口腔医学领域,特别涉及一种近红外比率型ph荧光探针及其制备方法与其在破骨吸收成像中的应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.正畸牙移动是通过对牙齿施加一定的力量,促使牙齿周围牙周膜及牙槽骨组织发生改建,最终实现牙齿空间位置的变化。在合适大小的正畸力作用下,受压侧牙周膜在压力的作用下会产生白细胞介素-1(interleukin-1,il-1)、前列腺素和il-6等促炎因子,促进破骨细胞分化成熟,随后成熟破骨细胞在牙槽骨表面进行破骨吸收活动,牙齿位置发生改变。在较大正畸力作用下,受压侧牙周膜组织因过度受压会形成局部坏死透明样变组织,此时牙齿移动速度会减慢;随后透明样变组织释放各类趋化因子及炎症因子促进破骨细胞分化成熟,发生潜掘性破骨吸收对坏死组织进行吸收,在该过程中临近坏死透明样变组织的牙槽骨会发生破坏性吸收,同时可能伴有一定程度牙根外吸收。因此提示检测牙移动过程中的破骨吸收活动对可控的健康牙移动具有重要意义。
4.目前针对正畸牙移动破骨吸收的检测主要有两种方式,一种是免疫组织化学技术检测,通过对所构建的正畸加力牙移动动物模型进行组织切片后,利用免疫组织化学技术进行染色检测;另一种则是微型计算机断层成像技术(micro computed tomography,micro ct),利用micro ct对加力前后牙槽骨骨密度、骨体积、骨小梁厚度等结构变化指标进行检测,以反映体内破骨吸收情况。免疫组织化学技术检测需要进行切片处理后方可对破骨吸收情况进行检测分析;micro ct技术则需通过对牙槽骨骨组织结构和矿物含量变化的评估来反映牙移动破骨吸收情况,其无法检测到与牙槽骨组织结构变化相关的破骨吸收活动。研究表明骨组织结构变化是滞后于体内实际破骨细胞骨吸收活动的,在牙槽骨组织形态发生显著变化前通过免疫组化染色可以检测到破骨细胞活动的上调。目前尚未有有效的检测方法可以实现对牙移动过程中破骨吸收活动的准确检测,从而能够在牙槽骨组织形态发生明显变化前及时提供真实有效的破骨吸收活动信息。
技术实现要素:
5.为了解决上述问题,本发明提供一个基于半花菁类结构的近红外比率型ph荧光探针,将其应用于正畸牙移动过程中破骨吸收活动的检测,为后续正畸牙移动过程中破骨吸收相关因素研究提供更为直观、准确的检测方法。
6.为了实现上述目的,本发明采用如下技术方案:
7.本发明的第一个方面,提供了一种用于破骨吸收成像的近红外比率型ph荧光探
针,结构式如式ⅰ所示:
[0008][0009]
本发明的第二个方面,提供了上述的以天冬氨酸六肽为骨靶向基团,用于破骨吸收成像的近红外比率型ph荧光探针的构建方法及骨靶向验证,包括:
[0010]
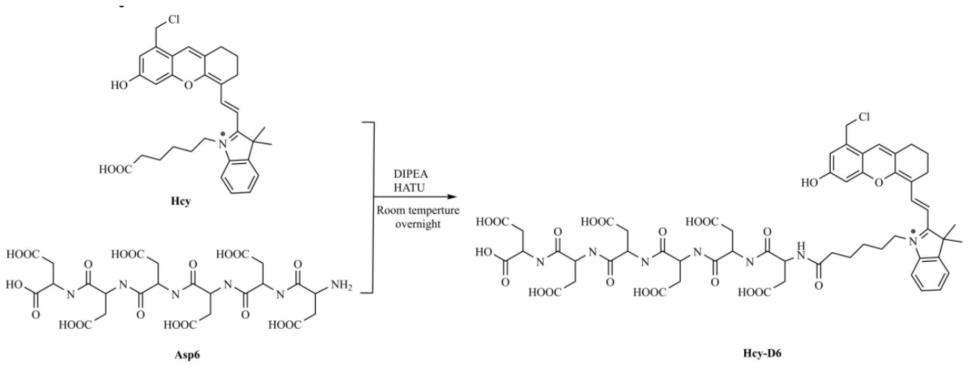
将n,n-二异丙基乙胺dipea与2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯hatu溶于二甲基亚砜dmso中,加入半花菁结构hcy,混合均匀,加入天冬氨酸六肽asp6进行反应,即得。
[0011]
羟基磷灰石(ha)结合实验验证骨靶向能力:将hcy-d6与hcy分别与ha结合,离心水洗后取沉淀物置于不同ph缓冲液中,于633nm激发波长下检测其在678nm及715nm处荧光信号。
[0012]
本发明的第三个方面,提供了一种体外破骨吸收检测方法,包括:
[0013]
利用密度梯度离心法纯化诱导骨髓单核细胞为破骨细胞,将其培养在无菌骨磨片上构建体外破骨吸收模型,甲苯胺蓝染色及扫描电镜检测骨吸收陷窝形成。随后将无菌骨磨片在hcy-d6溶液中孵育后,pbs清洗后置于24孔板内,将破骨细胞、成骨细胞分别按照前者每孔1
×
105,后者每孔1
×
104的细胞量均匀铺入含有hcy-d6预处理的无菌骨磨片的24孔板内,培养10~12天后,用hoechest33342对细胞核进行荧光染色示踪,随后将骨磨片置于激光共聚焦显微镜下在633nm的激发波长下检测其在678nm和715nm处荧光信号的变化,image j分析结果。
[0014]
本发明的第四个方面,提供了一种正畸加力牙移动过程中破骨吸收活动的直接检测方法,包括:
[0015]
在加力7天后对实验组及对照组进行体内成像检测,成像检测前3天,根据体重按照3.5mg/kg剂量在左侧上颌第一磨牙近中颊侧进行局部注射上述的hcy-d6;成像当天取出实验组及对照组左侧上颌骨,利用双光子激光共聚焦显微镜对左侧上颌第一磨牙近中根的近中颊侧牙槽骨进行荧光成像,成像条件:激发光为800nm,收集两个发射通道678
±
15nm及715
±
15nm的荧光信号,实验组和对照组所有成像条件设置均一致,图像处理由image j完成。
[0016]
本发明的有益效果
[0017]
(1)本发明以半花菁分子结构为主体,构建了一个近红外比率型ph荧光探针hcy-d6,hcy-d6利用分子内电荷转移原理可以实现对ph的比率性响应。本发明对hcy-d6的荧光性质、ph响应性进行了检测,结果表明该探针具有优秀的荧光特性,拥有两个最大发射峰(678nm/715nm),均位于近红外区域,有着良好的ph响应性:随着ph的升高最大发射峰会从678nm红移至715nm,为此可以有效的实现对ph变化的检测。此外,cck8细胞毒性检测及活死细胞染色实验证明该荧光探针具有较低的细胞毒性。
[0018]
(2)本发明在探针结构上引入天冬氨酸六肽结构,利用天冬氨酸六肽与骨组织表面羟基磷灰石组分结合的能力,成功将该荧光探针靶向输送至牙槽骨表面。甲苯胺蓝染色及扫描电镜检测显示成功构建体外破骨吸收模型;荧光检测结果显示破骨细胞组成功检测到678nm和715nm处的荧光信号变化,成骨细胞组未检测到相应荧光信号变化,证明hcy-d6可以检测出破骨细胞进行破骨吸收活动时的ph变化,同时hcy-d6的荧光信号变化不受成骨细胞活动影响。以上证明通过检测hcy-d6的荧光信号变化可以实现对破骨吸收活动的检测。
[0019]
(3)本发明利用hcy-d6成功实现对正畸牙移动破骨吸收活动的检测:荧光检测结果显示实验组荧光信号面积与对照组相比具有统计学意义的差异,说明加力7天后左侧上颌第一磨牙近中压力侧破骨吸收活动增多;ph半定量分析显示这两组的ph变化一致,均与以往破骨吸收ph变化范围相符,证明hcy-d6可以实现牙移动过程中破骨吸收活动的检测。
[0020]
(4)本发明制备方法简单、实用性强,易于推广。
附图说明
[0021]
构成本技术的一部分的说明书附图用来提供对本技术的进一步理解,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。
[0022]
图1为本发明实施例1中hcy-d6合成线路图;
[0023]
图2为本发明实施例1中探针结构及ph响应机理示意图;
[0024]
图3为本发明实施例1中hcy-d6的紫外-可见吸收光谱;
[0025]
图4为本发明实施例1中hcy-d6的荧光光谱及pka测定;
[0026]
图5为本发明实施例1中hcy-d6的光学稳定性及可逆性;
[0027]
图6为本发明实施例1中hcy-d6(10μm)的抗干扰性(
****
p<0.0001,ns:nosignificance);
[0028]
图7为本发明实施例1中不同浓度hcy-d6(0-200μm)与破骨细胞共培养24h后的细胞毒性性检测;a.cck-8法检测结果(
**
p<0.01,ns:no significance);b.活死细胞染色法检测结果(scale bar:50μm)。
[0029]
图8为本发明实施例2中羟基磷灰石(ha)与hcy-d6孵育后在不同ph缓冲液中的激光共聚焦图片(激发波长:633nm,cyan通道:678
±
15nm,red通道:715
±
15nm,bf:明场,scale bar:10μm)。
[0030]
图9为本发明实施例2中羟基磷灰石(ha)与hcy孵育后在不同ph缓冲液中的激光共聚焦图片(激发波长:633nm,cyan通道:678
±
15nm,red通道:715
±
15nm,bf:明场,scale bar:10μm)。
[0031]
图10为本发明实施例2中体外破骨细胞诱导及功能鉴定,a.诱导3天后的破骨前体细胞形态;b.诱导6天后的破骨细胞形态;c.诱导7天后trap染色示早期破骨细胞;d.诱导7天后trap染色示成熟破骨细胞;e.体外破骨细胞骨吸收陷窝的甲苯胺蓝染色;f.体外破骨细胞骨吸收陷窝的扫描电镜检查,e和f中的白色箭头所示即为骨吸收陷窝。
[0032]
图11为本发明实施例2中体外破骨吸收活动检测;a.成熟破骨细胞(oc)及成骨细胞(ob)培养在50μm hcy-d6预处理的无菌骨磨片上的激光共聚焦荧光图像;b.体外破骨吸收最大发射峰荧光强度比值的统计分析(激发波长:633nm;发射波长:cyan(678
±
15nm),
red(715
±
15nm))。
[0033]
图12为本发明实施例3中大鼠正畸牙移动模型构建;a.大鼠正畸牙移动模型构建示意图;b.加力7d后口内照片。
[0034]
图13为本发明实施例3中大鼠正畸牙移动破骨吸收荧光成像,a.对照组(0d)左侧上颌第一磨牙近中根的近中颊侧牙槽骨组织的三维荧光图像重建;b.实验组(7d)左侧上颌第一磨牙近中根的近中颊侧牙槽骨组织的三维荧光图像重建;c.对照组(0d)和实验组(7d)荧光面积统计学分析(
****
p<0.0001)(激发波长:800nm;发射波长:绿光通道(678
±
15nm),红光通道(715
±
15nm))。
[0035]
图14为本发明实施例3中大鼠正畸牙移动破骨吸收荧光强度比值分析,a.对照组(0d)和实验组(7d)的荧光成像图片;b.对照组(0d)和实验组(7d)荧光图像的最大发射峰荧光强度比值(i
678
/i
715
)的统计分析;c.对照组和实验组破骨吸收ph半定量分析(ns:no significance)(激发波长:800nm;发射波长:绿光通道(678
±
15nm),红光通道(715
±
15nm))。
具体实施方式
[0036]
应该指出,以下详细说明都是例示性的,旨在对本技术提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本技术所属技术领域的普通技术人员通常理解的相同含义。
[0037]
下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
[0038]
以下实施例中,采用的实验试剂如下:
[0039]
n,n-二异丙基乙胺(n,n-diisopropylethylamine,dipea)、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(2-(7-azabenzotriazol-1-yl)-n,n,n',n'-tetramethyluronium hexafluorophosphate,hatu)、二甲基亚砜(dimethyl sulfoxide,dmso)、乙醚、色谱纯乙腈均购置于国药试剂集团;nacl、cacl2、kcl、mgcl2、zncl2、fecl3、fecl2、cucl2、alcl3、naoh、hcl、柠檬酸、磷酸氢二钠磷酸二氢钠等均购置于阿拉丁试剂公司;cck-8活性检测试剂盒购于日本同仁公司;live/dead细胞活力/细胞毒性试剂盒购于美国invitrogen公司、磷酸盐缓冲液(phosphate buffered saline,pbs)(biosharp,美国)、双抗(100u/ml青霉素 100ug/ml链霉素)(hyclone,美国)、α-mem培养基(alpha modified eagle medium)(hyclone,美国)、胎牛血清(fetal bovine serum,fbs)(gibco brl,美国)、histopaque-1083细胞分离液(sigma,美国)、重组大鼠源性核因子κb受体活化因子配体(receptor activator of nf-κb ligand,rankl)(peprotech,美国)、重组大鼠源性巨噬细胞集落刺激因子(macrophage colony-stimulating factor,m-csf)(peprotech,美国)、抗酒石酸酸性磷酸酶染色试剂盒(tartrate resistant acid phosphatase,trap)(索莱宝,北京)、甲苯胺蓝染液(索莱宝,北京)、细胞培养瓶,离心管,细胞培养孔板(corning,美国)、异氟烷(瑞沃德,中国)、4%多聚甲醛(biosharp,美国)、羟基磷灰石(hydroxyapatite,ha)(拜阿蒙生物活性材料有限公司,中国)、异氟烷(瑞沃德,中国)、镍钛拉簧(tomy,日本)、不锈钢结扎丝(奥索医疗,中国)、卡瑞斯玛光固化树脂(贺利氏,德国)、牙科酸蚀剂(贺利氏,德国)。
[0040]
实施例1近红外比率型ph荧光探针合成及性质检测
[0041]
1.近红外比率型ph荧光探针hcy-d6构建,包括以下步骤:
[0042]
半花菁结构(hcy)及天冬氨酸六肽(aspartic acid hexapeptide,asp6)按照上图1所示的合成线路图,将dipea(78.86μl,2eq)与hatu(82.29mg,1.2eq)溶于2ml dmso中,加入hcy(120mg,1eq),室温搅拌2min后加入asp6(204.43mg,3eq),室温避光搅拌过夜。乙醚沉降除去dmso,随后将所得混合物用制备型hplc分离纯化,纯化条件:c18柱(5μm,20
×
250nm),流动相a为含0.01%甲酸的纯水,流动相b为色谱纯乙腈,按照20%a到100%b的梯度浓度以15ml/min流速洗脱45min,在波长610nm处检测吸光度。所得纯化产物用旋转蒸发仪(37℃)除去乙腈后,真空冻干干燥得到终产物hcy-d6(25mg,产率:9.1%)。1h nmr(400mhz,dmso)δ=8.82(s,1h),8.60(d,j=8.3,1h),8.18(s,1h),8.02(d,j=13.5,1h),7.57(d,j=8.0,1h),7.43(d,j=7.5,1h),7.28(s,2h),7.10(s,1h),7.04(s,2h),6.37(s,1h),6.15(s,1h),5.82(s,1h),5.77(s,3h),4.17(s,1h),3.89(s,4h),3.04(s,1h),2.98(s,6h),2.72(s,3h),2.64(s,4h),1.92(s,3h),1.84(s,2h),1.64(s,9h),1.50(s,3h),1.34(s,2h),1.22(s,1h).ftms(esi)m/z 1222.38865[m ].
[0043]
hcy-d6结构及ph响应机理如图2所示,当hcy-d6位于酸性环境时其结构上的酚羟基发生质子化,而位于碱性环境时酚羟基则发生去质子化,基于分子内电荷转移原理hcy-d6的荧光光谱发生改变,因此可以实现对ph的比率型响应。
[0044]
2.hcy-d6性质检测:
[0045]
hcy-d6溶于10%dmso溶液(纯水与dmso体积比为90:10),制成终浓度为1mm用于储存。用柠檬酸/磷酸氢二钠体系及0.1m盐酸和0.1m氢氧化钠配置ph为2.18-9.14的系列ph缓冲液用于后续检测。
[0046]
1)荧光性质检测
[0047]
利用ph为2.18-9.14的ph缓冲液配置终浓度为10μm的不同ph的hcy-d6检测溶液,将其置于1cm光程的石英比色皿中,然后利用u-4100分光光度计对位于不同ph缓冲液中的hcy-d6的紫外吸收光谱进行检测。利用f-4700荧光分光光度计在激发和发射的窄缝宽度设定为5nm,激发波长为658nm的条件下对不同ph缓冲液中的hcy-d6的荧光光谱进行扫描。结果如图3及图4所示,图3中a所示hcy-d6在不同ph的缓冲液中呈现不同颜色,在ph 2.18的溶液中呈现蓝色,在ph 9.14的溶液中则呈现青色。从图3中b显示的hcy-d6的紫外吸收光谱图中,可以看到hcy-d6在不同ph缓冲液中具有两个最大吸收峰,当其位于酸性溶液时最大吸收峰位于606nm处,随着ph的升高,其最大吸收峰向近红外区域发生了移动,由606nm变为690nm。图4中a所示,hcy-d6具有两个最大发射峰(678nm/715nm),均位于近红外区域(>650nm),在ph为2.18的缓冲液中其最大发射波长为678nm,随着ph的升高,其在678nm处的发射强度逐渐降低,在715nm处发射强度逐渐升高,当ph升高至5.9以上时其最大发射波长变为715nm,以上hcy-d6在不同ph缓冲液中最大发射峰的变化提示,它可以实现对ph的比率型检测。以最大发射峰荧光强度比值(i
678
/i
715
)为纵坐标,ph为横坐标作图,结果如图4中b所示,随着ph升高,i
678
/i
715
比值逐渐降低。随后对ph 5.0-6.5范围内最大发射峰荧光强度比值(i
678
/i
715
)与ph的线性关系进行了分析,如图4中c所示在ph 5.0-6.5的范围内i
678
/i
715
与ph具有很好的线性关系,相关系数为0.99。hcy-d6的pka值为5.92。
[0048]
2)稳定性及可逆性检测
[0049]
(1)光稳定性
[0050]
配置ph分别为2.24及6.86的缓冲液,加入10μm hcy-d6,连续2h内每5min用f-4700荧光分光光度计检测1次hcy-d6在658nm激发下的荧光信号变化,最后绘制以时间为横坐标,荧光信号强度比值为纵坐标的光稳定性图。结果如图5中a所示,在连续2h的检测中hcy-d6在ph 2.24及6.86的缓冲液中都表现出较好的稳定性,其荧光强度并没有随着激发次数增多而减弱,表明hcy-d6具有较强的抗光漂白能力,适用于连续追踪ph的变化。
[0051]
(2)可逆性
[0052]
利用0.1m hcl和0.1m naoh调节hcy-d6缓冲液(终浓度10μm)的ph为5.40或6.50,每隔10min调节1次ph,循环调节4次,用f-4700荧光分光光度计检测荧光信号变化。结果如图5中b所示,每次调节溶液ph后hcy-d6的荧光信号也随着ph的变化而快速变化,说明hcy-d6能够对h
快速响应;同时这两个荧光信号在这4次循环中都保持了稳定,说明hcy-d6具有较好的稳定性。以上结果说明hcy-d6对h
有着快速而稳定的响应,可以用于体内复杂而连续的ph变化检测。
[0053]
3)抗干扰性检测
[0054]
室温下,配置ph分别为2.86/7.52的含不同干扰物质的系列缓冲液:nacl(1mm)、cacl2(1mm)、kcl(1mm)、mgcl2(200μm)、zncl2(200μm)、fecl3(200μm)、fecl2(200μm)、cucl2(200μm)、alcl3(200μm),加入hcy-d6,终浓度为10μm,利用f-4700荧光分光光度计分别检测其发射光谱变化。结果如下图6所示,在ph为2.86和7.52的这两组实验中,在同一ph值的组内,即使存在不同阳离子,hcy-d6的荧光强度比值与对照组相比也并未存在统计学差异;只有在组间即同一阳离子的不同ph溶液中hcy-d6的荧光强度比值才发生了显著改变。以上表明hcy-d6的荧光信号变化只取决于h
,而对其他阳离子具有较强抗干扰性,可适用于破骨吸收过程中ph变化的检测。
[0055]
4)细胞毒性检测
[0056]
大鼠源性破骨细胞诱导培养见实施例2,经细胞计数后按照每孔3
×
103的细胞量将大鼠源性骨髓单核细胞均匀铺入96孔板内,每组3个复孔,一共铺2块板,每孔加入100μl含30ng/ml m-scf和50ng/ml rankl的含10%fbs的α-mem细胞培养液,每隔3天更换一次培养液,一共诱导7天。7天后诱导出成熟大鼠源性破骨细胞后,弃去原培养液,pbs清洗3次,每孔加入100μl不同浓度hcy-d6溶液(0/5/10/20/50/100/200μm),共培养24h后分别进行cck-8检测及活死细胞染色检测。结果如图7所示,a为共培养24h后的cck-8检测结果,可见即使浓度高达100μm,细胞存活率也在80%以上,只有浓度达到200μm时破骨细胞的细胞存活率才有所影响;b为共培养24h后的活死细胞染色,结果显示0-100μm hcy-d6与破骨细胞共培养24h后破骨细胞的存活率均较高,死细胞较少。上述结果说明了hcy-d6对破骨细胞的毒性较低,可以进行后续细胞实验。
[0057]
由此可知,本实施例合成了一个基于半花菁结构的近红外比率型ph荧光探针hcy-d6,对其光学性质的检测结果显示其具有两个最大强度发射峰678nm及715nm,均位于近红外区域,同时这两个发射峰的发射强度变化与ph具有较强的对应性,即随着ph升高其在678nm处的发射强度逐渐降低,在715nm处发射强度逐渐升高;hcy-d6在ph=5.0-6.5的范围内具有很好的灵敏度,此外还具有优秀的光稳定性和可逆性、较强的抗干扰能力以及较低的细胞毒性,充分表明hcy-d6适用于检测破骨吸收活动的ph变化。
[0058]
实施例2体外破骨吸收检测
[0059]
1.hcy-d6骨结合能力评估
[0060]
取1mg ha制成1ml 1mg/ml溶液,加入10μl 1mm hcy-d6或者hcy溶液(终浓度为10μm),37℃恒温摇床孵育1h。1h后,3000rpm离心5min去除上清,水洗离心2次,取沉淀物置于共聚焦小皿中,分别加入400μl ph为4-7的缓冲液,然后利用lsm780倒置激光共聚焦显微镜在633nm的激发波长下检测其在678nm和715nm处荧光信号的变化。结果如下图8及图9所示,hcy-d6组的ha在633nm激发光激发下可以检测到在678nm和715nm处明显的荧光信号,而hcy组的ha在相同激发条件下在678nm和715nm处并未检测到荧光信号,这说明hcy-d6利用其结构中的asp6成功与ha结合,随后在633nm激发光激发时结合在ha的hcy-d6发出荧光信号,因此ha可检测到荧光信号;而hcy结构上没有asp6导致其无法与ha结合,因此对该组ha进行荧光成像时无法检测到荧光信号。
[0061]
2.大鼠源性破骨细胞诱导及功能鉴定
[0062]
1)从山东大学动物实验中心购买5周龄雄性wistar大鼠5只,过量异氟烷吸入麻醉处死,置于75%酒精中消毒10min,将其转移至超净台中。无菌手术器械剪去双侧后肢表面皮肤及骨骼肌肉,在不损坏干骺端的前提下取出双侧股骨和胫骨,充分除去表面软组织,置于含2%双抗的pbs中充分冲洗。新的无菌手术剪剪去骨干两端骨骺,充分暴露髓腔,用5ml一次性注射器吸取含2%双抗的pbs,将其插入骨髓腔内清洗直至发白。收集骨髓腔清洗液,1500rpm离心5min后弃上清,用含15%fbs的α-mem细胞培养液重悬,加入m-csf(终浓度10ng/ml),置于25cm2培养瓶中,37℃、5%co2培养箱内培养24h。
[0063]
2)24h后,小心收集培养瓶内上层细胞悬液至15ml离心管内,1500rpm离心5min后弃去上清,加入5ml pbs重悬细胞。另取1个新的15ml离心管,加入等体积的histopaque-1083分离液,将pbs细胞重悬液缓慢加入分离液中,不破坏其液面分界面。室温条件下以400
×
g离心30min,离心结束取出离心管可见液体分层,其中位于中间的呈云雾状的细胞层即为本发明所需的骨髓单核细胞。小心去除单核细胞层上方液体,吸取云雾状单核细胞层液体转移至新的15ml离心管内。加入10ml pbs,充分混匀后,室温下250
×
g离心10min,弃去上清,pbs重悬,继续重复2次上述离心步骤以充分去除残留在单核细胞中的淋巴细胞分离液。随后用含10%fbs的α-mem细胞培养液重悬,细胞计数仪计数后,以每孔1
×
105个细胞密度均匀铺入24孔板内,每孔加入0.5ml含10%fbs的α-mem细胞培养液,同时加入50ng/ml rankl及30ng/ml m-csf,以诱导其向破骨细胞分化。一共诱导7天,每3天换一次培养液。
[0064]
3)为了鉴定所纯化诱导的大鼠源性破骨细胞,对其进行了细胞trap染色鉴定以及破骨细胞功能鉴定。后者为将诱导的成熟破骨细胞接种在无菌牛股骨骨磨片上,继续培养3天后进行甲苯胺蓝染色及扫描电镜检查,以此检测所诱导的破骨细胞的骨吸收能力。无菌牛股骨骨磨片由新鲜牛股骨制备,利用硬组织切磨系统将牛股骨沿着长轴方向纵切为4cm
×
2cm,厚0.5mm的切片,用牙科高速涡轮机打磨至100μm厚度,修剪成2cm
×
2cm大小。超声波清洗机超声清洗3次,每次15min,120℃,0.4mpa高压灭菌后浸泡于含2%双抗的pbs溶液中置于4℃冰箱保存备用,用时将其置于24孔板内使用。
[0065]
4)trap染色结果如下图10中a和b所示,可以看到单核细胞经过3天的诱导后其细胞形态发生了改变,呈现多样化,大部分细胞出现细胞突起,体积增大,呈现出融合的趋势;诱导6天后细胞体积明显增大,同时细胞突起明显,局部可以观察到体积巨大、形态不规则
的多核细胞。随后对诱导7天后的破骨细胞进行trap染色鉴定,结果如下图10中c和d所示,细胞胞质内均出现了紫红色沉淀,呈现trap阳性,即经过7天诱导单核细胞成功分化为破骨细胞。
[0066]
5)成熟破骨细胞在无菌骨磨片上继续培养3天后的甲苯胺蓝染色结果如图10中e所示,可以看到在骨磨片上存在局部点状、条索状不规则蓝紫色染色区域(白色箭头所示位置),即为骨吸收陷窝。
[0067]
6)成熟破骨细胞在无菌骨磨片上继续培养3天后的扫描电镜检查结果如图10中f所示,可以看到在骨磨片表面存在不同吸收深度的吸收陷窝(白色箭头所示区域),说明本研究所纯化诱导出来的破骨细胞具有良好的骨吸收能力,能对骨磨片表面骨组织进行吸收降解形成骨吸收陷窝。
[0068]
3.体外破骨吸收活动检测
[0069]
无菌骨磨片在50μm的hcy-d6溶液中于37℃条件下孵育1h,pbs清洗2遍后置于24孔板内。大鼠骨髓源性单核细胞分离纯化同前,将分离纯化的大鼠源性单核细胞及大鼠源性成骨细胞经细胞计数后按照前者每孔1
×
105,后者每孔1
×
104的细胞量均匀铺入含有hcy-d6预处理的无菌骨磨片的24孔板内,每组3个复孔。每3天更换一次培养液,破骨细胞诱导培养液为含30ng/ml m-scf和50ng/ml rankl的含10%fbs的α-mem细胞培养液,大鼠源性成骨细胞培养液则为含10%fbs的α-mem细胞培养液。一共培养10天后,用hoechest 33342对细胞核进行荧光染色示踪,随后将骨磨片置于激光共聚焦显微镜下在633nm的激发波长下检测其在678nm和715nm处荧光信号的变化,image j分析结果。结果如下图11中a所示,可以看到在破骨细胞组(oc)可以在细胞周围检测到678nm和715nm处明显的荧光信号变化,而在成骨细胞组未检测到相关荧光信号。随后对破骨细胞组荧光信号强度比值进行了分析,结果显示其荧光强度比值均值在1.458附近,根据第一部分实验中所测得的hcy-d6最大发射峰荧光强度比值与ph之间的关系对其进行半定量分析可知该局部ph在5.47左右。
[0070]
由此可知,本发明通过对大鼠源性骨髓单核细胞进行纯化诱导,成功培养出高纯度的破骨细胞,将成熟破骨细胞接种在牛股骨磨片上形成了明显的骨吸收陷窝,证明本发明所培养的破骨细胞具有较好的破骨吸收能力。随后利用hcy-d6对培养在骨磨片上的成骨细胞及破骨细胞的细胞活动进行检测,在破骨细胞组检测到明显的荧光信号变化,对该荧光信号变化进行半定量分析显示与以往研究的破骨吸收ph变化范围相符;而成骨细胞组未检测到荧光信号变化。以上充分说明hcy-d6的荧光信号变化只与破骨吸收过程中的ph下降有关,而不受成骨细胞活动影响,提示本发明可以通过检测hcy-d6的荧光信号变化来实现对破骨吸收活动的检测。
[0071]
实施例3正畸牙移动破骨吸收检测
[0072]
本实验经山东大学口腔医学院动物伦理委员会批准后进行(伦理审查批件:no.20201104),于山东省口腔组织再生重点实验室的动物实验平台完成相应实验操作。于济南朋悦实验动物繁育有限公司购买体重为250-280g的7周龄无特异病原菌动物级别健康雄性wistar大鼠,将其饲养于ivc动物饲养系统中,按12h周期昼夜更替,给予清洁饲料和凉开水,定期更换垫料,将其分为实验组和对照组两组,每组5只大鼠,实验组构建正畸牙移动模型,对照组则不进行加力处理,其余条件均与实验组相同。
[0073]
1.大鼠正畸牙移动模型构建
[0074]
所购买的10只wistar雄性大鼠经适应性饲养1周后随机分为实验组和对照组两组进行后续实验。实验组大鼠置入密封麻醉箱内用4%异氟烷进行诱导麻醉,待其意识消失后后将其转移至手术台,以仰卧位置于手术台上,固定四肢,利用自制开口器暴露手术视野,于左侧上颌第一磨牙处建立模型,术中利用面罩输入4%异氟烷及2l/min氧气混合气体用于维持麻醉,在麻醉状态下进行实验操作。利用不锈钢结扎丝穿过左侧上颌第一磨牙远中在近中处与镍钛拉簧相连,拉簧的另一端则由不锈钢结扎丝固定于上颌中切牙上用高速涡轮机磨出来的定位沟上,为防止脱落,在上颌中切牙处利用牙科酸蚀剂进行处理后用光固化树脂将结扎丝与中切牙固定在一起,从而加强固位。建模过程中镍钛拉簧的力量通过精细测力计控制在25g的力值上。上述手术操作完成后密切观察大鼠呼吸情况直到完全清醒,术后给予粉状饲料进食,以防止咀嚼较硬食物导致加力装置脱落。每日观察加力装置及口内情况,如有松动或者脱落及时进行加固处理。对照组不进行模型构建处理,其余饲养条件则与实验组相同。结果如图12所示,图12中a所示为大鼠正畸牙移动模型示意图,在左侧上颌第一磨牙处构建加力模型,以上颌中切牙为支抗,通过镍钛拉簧施加25g的拉力,使其近中移动。从图12中b中可以看出经过7天加力后,左侧上颌第一磨牙发生近中移动。
[0075]
2.正畸牙移动破骨吸收检测
[0076]
在加力7天后对实验组及对照组进行体内成像检测,成像检测前3天,根据体重按照3.5mg/kg剂量在左侧上颌第一磨牙近中颊侧进行局部注射hcy-d6。成像当天取出实验组及对照组左侧上颌骨,利用双光子激光共聚焦显微镜对左侧上颌第一磨牙近中根的近中颊侧牙槽骨进行荧光成像,成像条件:激发光为800nm,收集两个发射通道(678
±
15nm及715
±
15nm)的荧光信号,实验组和对照组所有成像条件设置均一致。图像处理由image j完成。结果如图13所示,可以看到加力7天后牙槽骨组织内荧光信号分布明显比对照组0天的广。利用image j软件可对这两组荧光面积进行测量,随后对所得数据进行独立t检验分析,结果如图13中c所示加力7天后大鼠左侧上颌第一磨牙近中颊侧牙槽骨组织内荧光面积与对照组0天的相比具有统计学意义的差异(p<0.0001)。
[0077]
随后本发明对对照组和实验组的荧光检测结果进行了荧光信号强度比值分析,结果如下图14所示,绿色荧光信号为最大发射峰678nm处荧光信号,红色荧光信号为最大发射峰715nm处荧光信号,a为对照组和实验组在678nm和715nm处的荧光信号检测结果,从这里也可以看出来加力7天后牙槽骨组织内的荧光信号明显要比对照组多。b则是对所有对照组和实验组的荧光图像的最大发射峰荧光强度比值(i
678
/i
715
)的统计分析,结果显示对照组最大发射峰荧光强度比值均值为1.344,实验组的是1.293,对这两组数据进行独立t检验,显示这两组数据没有统计学差异(p>0.05)。根据实施例1中hcy-d6最大发射峰荧光强度比值与ph之间的关系对实验组和对照组的破骨吸收ph变化进行半定量分析,结果显示实验组破骨吸收的ph均值为5.668,而对照组破骨吸收ph均值为5.635,独立t检验显示这两组数据没有统计学差异(p>0.05)。
[0078]
本实施例中,体内荧光成像结果显示在对照组和实验组均能检测到hcy-d6的荧光信号变化,与对照组相比,加力7天后牙槽骨内荧光信号明显增多,说明加力7天后的牙槽骨中破骨吸收活动较为活跃。同时对这两组的破骨吸收的ph变化进行半定量分析显示实验组与对照组破骨吸收ph变化是一致的,且与以往研究相符合,充分证明了本研究所合成的探针hcy-d6可以实现对牙移动破骨吸收活动的检测。
[0079]
以上所述仅为本技术的优选实施例而已,并不用于限制本技术,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。