1.本发明涉及天然药物提取分离领域,具体涉及一种天然鞘磷脂的制备方法。
背景技术:
2.鞘磷脂(sphingomyelin,sm)存在于大多数哺乳动物细胞膜内,是构成生物膜的重要成为之一,sm及其代谢产物具有多种生物活性,调节细胞功能。作为重要的信号分子参与调节细胞的生长、分化、衰老和凋亡。近年来,经过大量的学者研究,发现鞘磷脂及其代谢产物调控疾病发展的多个环节。如:位于细胞膜、膜蛋白的鞘磷脂可调节生长因子受体和超细胞基质蛋白的活动,其代谢产物可调节细胞分化、调节细胞免疫、参与调节炎症反应、诱导部分癌细胞凋亡等。此外,有学者指出,鞘磷脂作为一种潜在的功能食品,具有降低血清胆固醇、改善皮肤屏障功能和促进婴幼儿神经发育等作用。
3.鞘磷脂在蛋黄卵磷脂中是极微量的存在,一般在3%以下,想要分离出高纯度的鞘磷脂是及其困难的。又因其含量极小,在分离精制过程中就要尽量避免不必要的损失,提高精制收率。因此,如何在避免不必要的损失下高效制备出高纯度的鞘磷脂具有重要的意义。
4.目前制备高纯度的鞘磷脂的方法主要是以蛋黄磷脂为原料,经过的硅胶柱层析分离。由于卵磷脂中磷脂种类繁多,通常选用极性相对较小的洗脱剂先洗脱出极性较小的磷脂,而磷脂酰胆碱与鞘磷脂极性比较接近,仅一次柱层析难以直接得到高纯度的鞘磷脂。而且,每次柱层析往往需要几十倍柱体积的溶剂,每次柱层析能获得高纯度鞘磷脂收率不到10%,这极大增加了鞘磷脂的制备成本,市售价格也是达到了上千元/克,因此,开发一条成熟的鞘磷脂制备工艺迫在眉睫。
技术实现要素:
5.本发明的目的在于,解决现有技术缺少成熟获取天然鞘磷脂的方法的问题。
6.为了解决上述技术问题,本发明采用如下技术方案:
7.一种天然鞘磷脂的制备方法,包括如下步骤:
8.a、得到鞘磷脂粗品;
9.b、将所述鞘磷脂粗品溶解在醇-醚-水三元混合溶液中,再进行重结晶,得到鞘磷脂产品;所述醇-醚-水三元混合溶液中包括短链醇、醚类溶剂和水。
10.重结晶作为一种常用的精制手段运用于各种化合物的纯化,但目前未见使用该方法用于提纯鞘磷脂。其主要原因在于,鞘磷脂通常混杂在大量的卵磷脂当中,而由于卵磷脂具有表面活性剂的功能,不同含量的鞘磷脂粗品在各溶剂中的溶解度均不同。在鞘磷脂含量较低时,短链醇类、醚类、卤代烷烃和正己烷等多种溶剂均能对其溶解。然而,由于这种溶解是因为卵磷脂的表面活性作用而带来的,因此在重结晶时,只有极少量的鞘磷脂能在卵磷脂不析出的前提下析出,而大量的鞘磷脂仍然会与卵磷脂裹挟着共同析出,造成鞘磷脂的大量损失,收率极低。因此常规溶剂与重结晶方法无法有效地分离鞘磷脂(sm)与卵磷脂(pm),难以用于鞘磷脂的精制。因此,需要寻找一种适合鞘磷脂结晶的溶剂体系。
11.本发明提供了一种天然鞘磷脂的制备方法,以蛋黄磷脂为原料,先使用极性相对较大的洗脱剂,直接分离出鞘磷脂粗品,再使用特定比例的三元混合溶剂对鞘磷脂粗品进行精制,制备出高纯度的鞘磷脂,并有较高的收率,分离效果较好。本发明的溶剂用量远低于反复的柱层析,易于产业化生产,极大的降低了鞘磷脂的制备成本。
12.优选的,步骤b中,所述醇-醚-水三元混合溶液中,所述短链醇体积用量与所述鞘磷脂粗品的质量比为2~10ml:1g,所述短链醇与所述醚类溶剂的体积比为1:2~8,所述水与所述短链醇的体积比为1:20~90。为保证磷脂混合物能全部溶解,且溶剂用量最少,优选地,短链醇为乙醇,进一步地,溶剂的体积与混合物质量比为2~4ml:1g。
13.优选的,步骤b中,所述醇-醚-水三元混合溶液中,所述短链醇包括甲醇、乙醇、丙醇、异丙醇中的一种或多种,所述醚类溶剂包括乙醚、丙醚、异丙醚中的一种或多种。
14.优选的,步骤b中,重结晶温度为0~6℃,重结晶次数为4~8次。
15.优选的,步骤b的具体操作为:将所述鞘磷脂粗品使用所述短链醇溶解,再加入所述醚类溶剂和所述水配置成所述醇-醚-水三元混合溶液,于低温下重结晶,经抽滤或离心后收集固体,干燥后得到所述鞘磷脂产品。
16.鞘磷脂粗品使用短链醇溶解,再加入醚类溶剂和水配置成三元混合溶液,有助于鞘磷脂粗品的溶解完全,并能提高溶解效率。如果先配置成三元混合溶液再用于溶解鞘磷脂粗品,则有造成样品难溶解、或者溶解不完全的情况出现,溶解效率也较低。
17.优选的,步骤b中,所述干燥包括减压干燥、真空干燥中的一种或多种。
18.优选的,步骤a的具体操作为:以蛋黄磷脂为原料,经过柱层析,得到鞘磷脂与磷脂酰胆碱混合物,即所述鞘磷脂粗品;所述鞘磷脂粗品中的鞘磷脂含量大于30%。
19.以者蛋黄来源的磷脂为原料,使用多卤代烷烃溶解,经柱层析以多卤代烷烃和短链醇的混合溶剂进行洗脱,得到鞘磷脂含量大于30%的混合物。再将上述混合物用短链醇溶解,加入其他两种溶剂制备成三元混合溶液,于低温下重结晶,最后经抽滤或离心得到高纯度鞘磷脂。
20.当鞘磷脂含量过低时,其物理溶解性会发生变化,因此,经柱层析所得鞘磷脂混合物中的鞘磷脂含量不得低于30%。
21.所用磷脂来源于者蛋黄。
22.优选的,步骤a中,所述柱层析为硅胶柱层析,硅胶与所述原料按照质量比4~8:1进行填料,为保证分离效果和减少溶剂用量,优选为6:1,采用多卤代烷烃-短链醇混合溶剂进行洗脱,所述多卤代烷烃-短链醇混合溶剂包括:多卤代烷烃、短链醇按体积比1:0.5~2混合,为更好分离磷脂酰胆碱和鞘磷脂,优选为1:0.5。
23.优选的,步骤a中,所述硅胶的100-200目,孔径为步骤a中,所述多卤代烷烃-短链醇混合溶剂中,所述多卤代烷烃包括二氯甲烷、三氯甲烷中的一种或多种。
24.一种如上述天然鞘磷脂的制备方法获得的鞘磷脂产品,所述鞘磷脂产品纯度高于98%。
25.与现有技术相比较,实施本发明,具有如下有益效果:
26.本发明技术方案以蛋黄来源的磷脂为原料,首先经过柱层析得到纯度高于30%以上的鞘磷脂,再使用三元混合体系的溶剂对鞘磷脂进行重结晶,经过减压干燥后得到纯度高于98%的天然鞘磷脂。该方法制备简单,溶剂用量少,精制收率高,极大降低了生产成本,
易于产业化生产。
附图说明
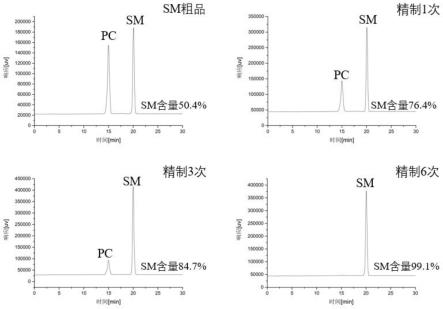
27.图1为实施例5鞘磷脂精制的效果图。
28.图2为效果例2精制一次的效果图。
具体实施方式
29.为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明作进一步地详细描述,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
30.实施例1
31.本实施例制备一种天然鞘磷脂的制备方法
32.s1:取10g蛋黄磷脂原料,加入20ml三氯甲烷溶解,填装50g硅胶进行柱层析,使用三氯甲烷-乙醇(1:0.5)进行洗脱,tlc监控,洗脱至sm斑点较明显时,开始收集样品,直至sm洗脱完全,减压浓缩得到sm粗品0.68g,纯度为41.3%。
33.s2:用3.4ml乙醇将sm粗品溶解,再加入0.06ml水和24ml乙醚混合均匀,将混合后的三相溶液置于0~6℃冰箱析晶,离心,除去上清液,得sm固体0.21g,纯度70.1%。加入1.1ml乙醇将sm固体溶解,再加入0.02ml水和7.7ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶4次,减压干燥,得sm纯品0.10g,纯度99.1%,精制收率(以sm计)为35.29%。
34.实施例2
35.本实施例制备一种天然鞘磷脂的制备方法
36.s1:取100g蛋黄磷脂原料,加入200ml二氯甲烷溶解,填装600g硅胶进行柱层析,使用二氯甲烷-甲醇(1:1)进行洗脱,tlc监控,洗脱至sm斑点较明显时,开始收集样品,直至sm洗脱完全,减压浓缩得到sm粗品6.75g,纯度为42.1%。
37.s2:用54ml甲醇将sm粗品溶解,再加入2ml水和270ml异丙醚混合均匀,将混合后的三相溶液置于0~6℃冰箱析晶,离心,除去上清液,得sm固体1.88g,纯度72.9%。加入7.6ml甲醇将sm固体溶解,再加入0.2ml水和60ml异丙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶4次,减压干燥,得sm固体0.97g,纯度98.7%,精制收率(以sm计)为33.69%。
38.实施例3
39.本实施例制备一种天然鞘磷脂的制备方法
40.s1:取500g蛋黄磷脂原料,加入1000ml三氯甲烷溶解,填装4000g硅胶进行柱层析,使用三氯甲烷-异丙醇(1:2)进行洗脱,tlc监控,洗脱至sm斑点较明显时,开始收集样品,直至sm洗脱完全,减压浓缩得到sm粗品35.1g,纯度为34.8%。
41.s2:用335ml丙醇将sm粗品溶解,再加入3.8ml水和2350ml丙醚混合均匀,将混合后的三相溶液置于0~6℃冰箱析晶,离心,除去上清液,得sm固体8.86g,纯度54.5%。加入70ml丙醇将sm固体溶解,再加入0.88ml水和490ml丙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶6次,真空干燥,得sm固体3.91g,纯度98.2%,精制收率(以sm计)为31.43%。
42.实施例4
43.本实施例制备一种天然鞘磷脂的制备方法
44.s1:取100g蛋黄磷脂原料,加入150ml三氯甲烷溶解,填装800g硅胶进行柱层析,使用二氯甲烷-乙醇(1:1)进行洗脱,tlc监控,洗脱至sm斑点较明显时,开始收集样品,直至sm洗脱完全,减压浓缩得到sm粗品6.1g,纯度为46.4%。
45.s2:用36ml异丙醇将sm粗品溶解,再加入0.4ml水和180ml乙醚混合均匀,将混合后的三相溶液置于0~6℃冰箱析晶,离心,除去上清液,得sm固体2.08g,纯度59.1%。加入18ml异丙醇将sm固体溶解,再加入0.2ml水和72ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶6次,真空干燥,得sm固体1.03g,纯度98.8%,精制收率(以sm计)为35.95%。
46.实施例5
47.本实施例制备一种天然鞘磷脂的制备方法
48.s1:取2kg蛋黄磷脂原料,加入4l三氯甲烷溶解,填装1.6kg硅胶进行柱层析,使用三氯甲烷-乙醇(1:0.5)进行洗脱,tlc监控,洗脱至sm斑点较明显时,开始收集样品,直至sm洗脱完全,减压浓缩得到sm粗品1040.8g,纯度为50.4%。
49.s2:取s1中sm粗品50g,用100ml乙醇溶解,再加入1.25ml水和200ml乙醚混合均匀,将混合后的三相溶液置于0~6℃析晶,离心,除去上清液,得sm固体30.18g,纯度70.4%。加入65ml乙醇将sm固体溶解,再加入0.8ml水和260ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶4次,减压干燥,得sm固体15.61g,纯度99.1%,精制收率(以sm计)为61.39%。
50.以本实施为代表,在不同精制次数下均进行相关物质含量的检测,结果如图1所示。从图1可以看出,随着精制次数的增加,sm的纯度得到了有效的提高。
51.实施例6
52.本实施例使用实施例5中的sm粗品进行制备
53.取实施例5中的sm粗品50g,用200ml乙醇将sm粗品溶解,再加入4ml水和1200ml乙醚混合均匀,将混合后的三相溶液置于0~6℃析晶,离心,除去上清液,得sm固体24.65g,纯度78.7%。加入100ml乙醇将sm固体溶解,再加入2ml水和600ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶3次,减压干燥,得sm固体15.13g,纯度99.4%,精制收率(以sm计)为59.68%。
54.实施例7
55.本实施例使用实施例5中的sm粗品进行制备
56.取实施例5中的sm粗品50g,用500ml乙醇将sm粗品溶解,再加入25ml水和4l乙醚混合均匀,将混合后的三相溶液置于0~6℃析晶,离心,除去上清液,得sm固体18.30g,纯度83.9%。加入160ml乙醇将sm固体溶解,再加入8ml水和1280ml乙醚混合均匀,置于0~6℃析晶,抽滤。再重复析晶3次,减压干燥,得sm固体10.59g,纯度99.2%,精制收率(以sm计)为41.69%。
57.对比例1
58.取实施例5中的sm粗品50g,用100ml乙醇将sm粗品溶解,加入400ml乙醚混合均匀,将混合后的两相溶液置于0~6℃析晶,离心,除去上清液,得sm固体17.16g,纯度65.2%。加入40ml乙醇将sm固体溶解,再加入160ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复
析晶3次,减压干燥,得sm固体4.12g,纯度98.9%,精制收率(以sm计)为16.17%。
59.对比例2
60.取实施例5中的sm粗品50g,用550ml乙醇将sm粗品溶解,再加入27.5ml水和2.2l乙醚混合均匀,将混合后的两相溶液置于0~6℃析晶,离心,除去上清液,得sm固体9.77g,纯度84.9%。加入110ml乙醇将sm固体溶解,再加入5.5ml水和440ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶3次,减压干燥,得sm固体3.14g,纯度99.3%,精制收率(以sm计)为12.37%。
61.对比例3
62.取实施例5中的sm粗品50g,用50ml乙醇将sm粗品溶解,再加入0.55ml水和100ml乙醚混合均匀,将混合后的两相溶液置于0~6℃析晶,离心,除去上清液,得sm固体31.7g,纯度60.1%。加入31ml乙醇将sm固体溶解,再加入0.35ml水和124ml乙醚混合均匀,置于0~6℃冰箱析晶,抽滤。再重复析晶3次,使用乙醇将固体溶解,减压干燥,得sm固体19.4g,纯度89.6%;再重复结晶3次,减压干燥,得sm固体15.71g,纯度91.4%,精制收率(以sm计)为56.98%。继续重复该精制操作,纯度未见明显提升。
63.对比例4
64.取实施例5中的sm粗品50g,加入500ml乙醚混合均匀,将混合后的浑浊液离心,除去上清液,得sm固体40.2g,纯度56.2%。再加入160ml乙醚混合均匀,浑浊液抽滤。再重复3次,减压干燥,得sm固体25.5g,纯度65.1%,精制收率65.88%,再重复3次,得sm固体19.5g,纯度69.1%,精制收率(以sm计)为52.7%。
65.对比例5
66.取实施例5中的sm粗品50g,用100ml乙醇将sm粗品溶解,再加入5ml水混合均匀,将混合后的两相溶液置于0~6℃析晶,未见晶体析出。在加入20ml水,置于0~6℃析晶,将混合后的浑浊液离心,除去上清液,得sm固体3.21g,纯度50.6%,可见无精制效果。
67.效果例1
68.从实施例1~7中可发现,运用本发明的方法,可以稳定得到纯度大于98%的鞘磷脂产品。另一方面,精制收率受粗品纯度的影响比较大。但即使是使用纯度较低的粗品,运用本发明精制后的收率仍基本维持在30%以上,相较于传统精制方法在收率方面也取得了显著的进步。本发明制备过程简单,溶剂用量少,极大降低了生产成本,易于产业化生产。
69.从实施例5~7中可发现,在权利要求限定范围内,乙醇比例越大,精制一次的纯度有所提高。实施例5与对比例2相比,乙醇的比例过大,则会造成sm析出难度加大,导致收率偏低。实施例5与对比例3相比,乙醇比例过低,易导致样品溶解不完全,造成磷脂酰胆碱和鞘磷脂混合析出,经过多次重结晶也难以达到98%以上纯度。
70.在短链醇中加适量的水相可降低醇对鞘磷脂和磷脂酰胆碱的溶解度,实施例5与对比例1相比,可以发现本发明使用短链醇、水、醚类组成的三元体系的精制收率(64.53%)远高于短链醇与乙醚组成的二元体系的收率(16.17%)。实施例5与实施例4相比,可以发现,仅加入醚类溶剂萃取磷脂酰胆碱,不仅会损失原料液中的鞘磷脂,其提纯程度有非常有限。通过对比例5,可发现仅使用短链醇-水体系无精制效果。可见,运用本发明的三元混合溶剂体系,可获得高纯度、高收率的鞘磷脂。
71.效果例2
72.取实施例5中的鞘磷脂粗品200g,平均分成4份,分别使用甲醇-水-乙醚、乙醇-水-乙醚、异丙醇-水-乙醚、丙醇-水-乙醚(按鞘磷脂粗品50g:醇500ml:水6.25ml:醚4l的比例)进行精制一次,使用甲醇-水-乙醚体系精制后纯度为79.6%,使用乙醇-水-乙醚体系精制后纯度为83.9%,使用异丙醇-水-乙醚体系精制后纯度为65.4%,使用丙醇-水-乙醚体系精制后纯度为62.7%。详细色谱图见图2。
73.在溶解度的测试中(鞘磷脂含量50.4%),甲醇与乙醇溶解度均大于2mg/ml,异丙醇醇、丙醇为溶解度均小于1mg/ml。而通过图2的对比发现,在用量相同情况下,甲醇-水-乙醚、乙醇-水-乙醚体系效果要优于异丙醇-水-乙醚、丙醇-水-乙醚体系,推测鞘磷脂粗品中的杂质更易溶于甲醇-水-乙醚、乙醇-水-乙醚体系,因此重结晶时不易被鞘磷脂包裹析出。
74.以上所揭露的仅为本发明的较佳实施例而已,当然不能以此来限定本发明之权利范围,因此依本发明权利要求所作的等同变化,仍属本发明所涵盖的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。