一种有机催化nazarov环化合成手性环戊烯酮类化合物的方法
技术领域
1.本发明属于有机合成领域,涉及手性环戊烯酮类化合物的合成,具体为有机催化nazarov环化合成手性环戊烯酮类化合物的方法。
2.
背景技术:
手性环戊烯酮是重要的有机合成中间体,并且广泛存在于天然产物及药物分子中(参见:a)roche,s.p.;aitken,d.j.eur.j.org.chem.2010,2010,5339-5358.b)simeonov,s.p.;nunes,j.p.m.;guerra,k.;kurteva,v.b.;afonso,c.a.m.chem.rev.2016,116,5744-5893;c)m.g.vinogradov,o.v.turova,s.g.zlotin,org.biomol.chem.2017,15,8245-8269.d)a.j.frontier j.j.hernandez,acc.chem.res.2020,53,1822-1832.)。目前合成手性环戊烯酮类化合物的最主要策略是使用1,4-二烯酮类化合物作为底物,经过nazarov环化反应得到(见:a)liang,g.;trauner,d.j.am.chem.soc.2004,126,9544-9545.b)he,w.;sun,x.;frontier,a.j.j.am.chem.soc.2003,125,14278-14279.c)cao,p.;deng,c.;
3.zhou,y.y.;sun,x.l.;zheng,j.c.;xie,z.;tang,y.angew.chem.,int.ed.2010,49,4463-4466;d)j.cao,m.-y.hu,s.-y.liu,x.-y.zhang,s.-f.zhu,q.-l.zhou.j.am.chem.soc.2021,143,6962-6968.等)。这种策略虽然发展比较成熟,但是该方法存在两个不足:第一:1,4-二烯酮类底物底物要求较高,往往需要有强给电子或吸电子基团,且反应条件为:金属催化,金属残留也是难于处理的问题。其二,1,4-二烯酮类环化形式比较单一,因此得到手性环戊烯酮种类比较有限,限制了手性环戊烯酮的多样性的开发和利用。因此,发展简洁高效的方法合成多样性手性环戊烯酮,特别是较复杂的手性环戊烯酮一直以来都是非常具有挑战的研究课题。
技术实现要素:
4.鉴于背景技术中存在的手性环戊烯酮种类多样性的不足,本发明提供了一种有机催化nazarov环化合成手性环戊烯酮类化合物的方法:以1,2-二酮化合物为原料,在绿色环保的手性硫脲催化剂的催化作用下,发生分子内nazarov环化,进而得到手性环戊烯酮类化合物。
5.为了实现本发明目的,所采用的技术方案为:
6.一种有机催化nazarov环化合成手性环戊烯酮类化合物的方法,包括步骤:在手性硫脲催化剂的催化作用下,1,2-二酮化合物发生分子内nazarov环化,得到手性环戊烯酮类化合物,结构式为
7.所述1,2-二酮底物的结构式为:
[0008][0009]
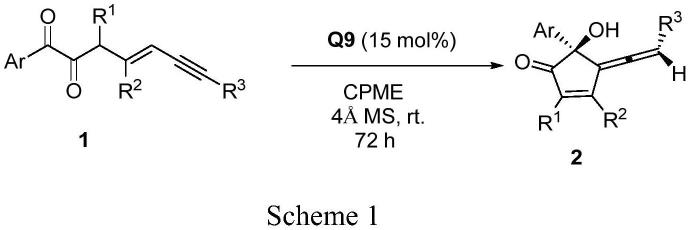
其中,ar为苯环上可带有吸电子或者给电子的含芳基基团;r1为甲基、乙基或者正丙基中的一种;r2为甲基、乙基或者苄基中的一种;r3为烷基(正丁基,叔丁基,环丙基等)或芳基。
[0010]
进一步的,所述方法按照下述步骤进行:在氩气保护下,向反应瓶中加入1,2-二酮底物,加入手性硫脲催化剂,或分子筛和溶剂(例如甲苯、二氯甲烷、二氯乙烷、环戊基甲醚中的任意一种或几种的组合),在室温下搅拌反应,待反应完全,淬灭、纯化,得到所述手性环戊烯酮类化合物。
[0011]
进一步的,手性硫脲结构式为:手性硫脲结构式中r为h或ome,r1为乙烯基或乙基。
[0012]
所采用的最优手性硫脲结构为,
[0013][0014]
具体的反应方程式如下(scheme 1):
[0015][0016]
进一步的,所述1,2-二酮的反应浓度为0.025-0.1m(更优选0.05m),即:1摩尔的反应底物对应2l溶剂。
[0017]
进一步的,所述硫脲催化剂用量为底物当量的10%-20%,当催化剂用量为15%时,可以达到最高收率和光学选择性。继续增大催化剂使用量,反应速率加快,收率和光学选择性不再增加。
[0018]
有益效果:
[0019]
本发明方法通过采用绿色环保的手性有机催化剂催化1,2-二酮底物制备一系列手性环戊烯酮化合物。该方法具有反应操作简单、条件温和,绿色环保的优点。同时,该方法制备的手性环戊烯酮化合物同时兼备碳手性中心以及联烯轴手性的特征。
[0020]
说明书附图
[0021]
图1为本发明实施例1中产物的hplc图;
[0022]
图2为本发明实施例1中产物的hplc图;
[0023]
图3为本发明实施例1中产物的hplc图;
[0024]
图4为本发明实施例1中产物的hplc图。
具体实施方式
[0025]
二酮底物1的制备方法:
[0026]
在氩气保护下,向反应瓶中加入1,4-烯炔醇、α-芳基重叠乙酸甲酯、醋酸铑及正己烷,在室温下反应。反应结束后,淬灭纯化,得到1化合物(见下图)。
[0027]
经试验验证,本技术手性环戊烯酮类化合物制备中所用化合物1的结构式中r1可以为甲基、乙基或者正丙基中的一种;r2可以为甲基、乙基或者苄基中的一种;r3可以为烷基或芳基,且所得手性环戊烯酮类化合物收率可达69~92%,立体选择性可达80~97%。下面结合实施例对本发明做进一步描述,但不限于此。
[0028]
实施实例1:
[0029]
(2a)的合成:
[0030][0031]
氩气保护下向反应管中加入1a(0.1mmol,32mg),环戊基甲醚(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(28.8mg,dr》19:1,收率:90%,95%ee)为淡黄色固体.熔点为165-168℃,对映选择性由高效液相色谱测定(hplc,手性柱:daicel chiralpak ic column;流动相:hexane/iproh=70:30,流速:1.0ml/min,λ=254nm,出峰时间:tr(major)=7.67min,tr(minor)=10.17min.95%ee.).[α]
d25
=-331.3(c=0.25in chcl3).1h nmr(400mhz,cdcl3)δ7.47-7.42(m,2h),7.34-7.25(m,3h),7.20-7.17(m,3h),6.94-6.92(m,2h),6.86(s,1h),3.23(s,1h),2.58-2.34(m,2h),2.17(s,3h),1.16(t,j=7.6hz,3h).
13
c nmr(100mhz,cdcl3)δ204.0,202.9,162.4,141.3,141.1,132.8,128.7,128.5,128.0,127.9,127.3,125.2,118.4,103.3,78.9,17.2,13.9,12.9.hrms(esi)calculated for c
22h21
o2[m h]
:317.1542,found:317.1544.(hplc图见图1)。
[0032]
基于本实施例,将分子筛等量替换为分子筛时结果:81%收率,90ee.,将分子筛等量替换为分子筛时结果:80%收率,88%ee.)。基于本实施例,还进行了手性硫脲催化剂适用性试验,结果显示以q9为催化剂(也即本实施例)效果最好,具体如下:
[0033][0034]
实施实例2:
[0035]
(2b)的合成:
[0036][0037]
氩气保护下向反应管中加入1b(0.1mmol,33mg),环戊基甲醚(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2b(27.6mg,dr》19:1,收率:84%,90%ee)为淡黄色固体.熔点为176-178℃,对映选择性由高效液相色谱(hplc,手性柱:daicel chiralpak ic column;流动相:hexane/iproh=70:30,流速:1.0ml/min,λ=254nm,出峰时间:tr(major)=8.64min,tr(minor)=11.22min)[α]
d25
=-102.7(c=0.1in chcl3).1h nmr(400mhz,cdcl3)δ7.31(d,j=8.0hz,2h),7.19-7.16(m,3h),7.09(d,j=8.0hz,2h),6.99-6.94(m,2h),6.83(s,1h),3.18(s,1h),2.52-2.36(m,2h),2.30(s,3h),2.13(s,3h),1.12(t,j=7.6hz,3h).
13
c nmr(100mhz,cdcl3)δ204.0,202.8,162.1,140.9,138.2,137.8,132.9,129.2,128.7,127.9,127.3,125.2,118.5,103.3,78.9,21.2,17.2,13.8,12.9.hrms(esi)calculated for c
23h23
o2[m h]
:331.1698,
found:331.1643.(hplc图见图2)。
[0038]
实施实例3:
[0039]
2c的合成:
[0040][0041]
氩气保护下向反应管中加入1c(0.1mmol,47mg),环戊基甲醚(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2b(32.0mg,dr》19:1,收率:68%,84%ee)为油状液体.对映选择性由高效液相色谱(hplc,手性柱:daicel chiralpak ia column;流动相:hexane/iproh=70:30,流速:1.0ml/min,λ=254nm,出峰时间:tr(major)=5.65min,tr(minor)=5.20min).[α]
d25
=-76.7(c=0.1in chcl3).1h nmr(400mhz,cdcl3)δ8.12(d,j=7.6hz,1h),7.60(s,1h),7.50(d,j=7.9hz,1h),7.33-7.18(m,1h),7.1-6.95(m,4h),6.85(s,1h),6.80(d,j=7.3hz,2h),3.14(s,1h),2.55-2.42(m,2h),2.19(s,3h),1.62(s,9h),1.15(t,j=7.6hz,3h).
13
c nmr(100mhz,cdcl3)δ203.1,202.6,161.7,149.5,140.3,136.3,132.5,128.6,127.9,127.5,127.4,124.6,123.8,122.7,120.7,120.6,116.7,115.4,103.3,83.9,76.5,28.3,17.3,13.9,12.9.hrms(esi)m/z calculated for c
29h29
nnao4[m na]
:478.1989,found:478.1991.(hplc图见图3)。
[0042]
实施实例4:
[0043]
2d的合成:
[0044][0045]
氩气保护下向反应管中加入1d(0.1mmol,30mg),环戊基甲醚(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2d(27.0mg,dr》19:1,收率:90%,86%ee)为淡黄色固体,熔点:86-88℃;对映选择性由高效液相色谱(hplc:手性柱:daicel chiralpak ic column;流动相:hexane/iproh=50:50,流速:1.0ml/min,λ=254nm,出峰时间:tr(major)=6.48min,tr(minor)=9.41min.86%ee).[α]
d25
=-65.7(c=0.2in chcl3).1h nmr(300mhz,cdcl3)δ7.46-7.06(m,5h),5.84(s,1h),3.02(s,1h),2.49-2.34(m,2h),2.11(s,3h),1.12(t,j=7.6hz,3h),0.81(s,9h).
13
c nmr(75mhz,cdcl3)δ204.6,197.8,163.8,141.5,139.9,128.3,127.7,125.1,116.7,112.1,78.3,33.8,30.0,17.1,13.6,13.0.hrms(esi)calculated for c
20h25
o2[m h]
:297.1855,
found:297.1850.(hplc图见图4)。
[0046]
实施例5
[0047]
2a的合成:
[0048]
氩气保护下向反应管中加入1a(0.1mmol,32mg),二氯甲烷(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(29.1mg,dr》19:1,收率:91%,71%ee)为淡黄色固体.
[0049]
实施例6
[0050]
2a的合成:
[0051]
氩气保护下向反应管中加入1a(0.1mmol,32mg),甲苯(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(25.2mg,dr》19:1,收率:80%,75%ee)为淡黄色固体.
[0052]
实施例7
[0053]
2a的合成:
[0054]
氩气保护下向反应管中加入1a(0.1mmol,32mg),环戊基甲醚(2.0ml),手性催化剂q9(10mol%,7.0mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(16.8mg,dr》19:1,收率:50%,95%ee)为淡黄色固体.
[0055]
实施例8
[0056]
2a的合成:
[0057]
氩气保护下向反应管中加入1a(0.1mmol,32mg),环戊基甲醚(2.0ml),手性催化剂q9(20mol%,14.0mg),分子筛(60mg),在室温下反应60小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(28.8mg,dr》19:1,收率:90%,95%ee)为淡黄色固体.
[0058]
实施例9
[0059]
2a的合成:
[0060]
氩气保护下向反应管中加入1a(0.1mmol,32mg),环戊基甲醚(1.0ml,浓度0.1m),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(25.8mg,dr》19:1,收率:80%,92%ee)为淡黄色固体.
[0061]
实施例10
[0062]
2a的合成:
[0063]
氩气保护下向反应管中加入1a(0.1mmol,32mg),环戊基甲醚(4.0ml,浓度0.025m),手性催化剂q9(15mol%,10.5mg),分子筛(60mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(18.8mg,dr》19:1,收率:60%,95%ee)为淡黄色固体.
[0064]
实施例11
[0065]
2a的合成:
[0066]
氩气保护下向反应管中加入1a(0.1mmol,32mg),环戊基甲醚(2.0ml),手性催化剂q9(15mol%,10.5mg),分子筛(30mg),在室温下反应72小时,待反应完全,加入氯化铵(10ml)淬灭,乙酸乙酯萃取(10*3ml)合并有机相,减压浓缩,粗产物通过柱层析纯化得到2a(27.0mg,dr》19:1,收率:88%,93%ee)为淡黄色固体.
[0067]
所述实施例为本发明的优选的实施方式,但本发明并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员能够做出的任何显而易见的改进、替换或变型均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。