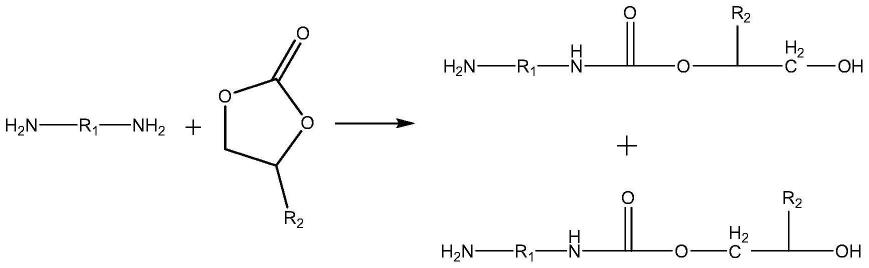
1.本发明属于化学制药领域,具体涉及一种高纯格列波脲的制备方法。
背景技术:
2.格列波脲又叫甲磺冰脲,商品名克糖利(glutril),属第二代磺脲类降糖药。克糖利作用强度中等,25mg疗效强于0.5g甲苯磺丁脲。对血中三酰甘油、胆固醇及磷脂均无明显影响,有降低血小板黏附和聚积、抗脂肪分解作用,有利于防治糖尿病血管并发症,适用于用于非胰岛素依赖型糖尿病,对其他磺脲类药物失效者仍可选用。另外磺胺、水杨酸、保泰松、双香豆素等可增强本品作用。
3.格列波脲为磺脲类降糖药,它与甲苯磺丁脲、格列吡嗪、格列齐特、格列本脲、格列美脲、格列喹酮等又称为“磺脲类促胰岛素分泌剂”,它们有共同的降糖作用机理,即与胰腺β细胞膜上的磺酰脲受体结合,从而启动胰岛素的分泌来达到降糖的目的,它们的降糖优点各异,但毒副作用都差不多,仅仅是程度的不同而已;另一方面在结构上它们都以苯磺酰脲为主要官能团,其中格列波脲与其他几个格列类药物的区别在于它们各自所包含的基团不同。
4.目前为止,关于格列波脲的合成报道还是非常少的,如何高效地实现格列波脲的合成将直接关系到格列波脲的质量控制以及用药的安全性,对于进一步研究该药物的生理活性、毒性以及纯度表征是至关重要的。
技术实现要素:
5.本发明的目的在于提供一种能够得到收率与纯度较高的格列波脲。
6.本发明为实现上述目的所采取的技术方案为:本发明的另一目的是一种高纯格列波脲的制备方法,其步骤包括:-由肟类中间体3在氢氧化钠、锌粉条件下制得氨基酮类中间体4;-上述氨基酮类中间体4经氢化铝还原制得氨基醇类中间体5;-上述氨基醇类中间体5在氢氧化钠、二氯甲烷条件下反应,经萃取、浓缩与柱色谱分离得到氨基甲酸酯类中间体6;-将上述氨基甲酸酯类中间体6在碱性条件下加热回流,得到氨基醇7,然后加入tsnco,经柱色谱分离得到格列波脲;tsnco,经柱色谱分离得到格列波脲;tsnco,经柱色谱分离得到格列波脲;。
7.本发明提供的一种高纯格列波脲,其纯度>99.5%。
8.本发明提供的格列波脲的合成路线反应条件温和,易于调控,生产周期短,设计合理,能够制得具有较高的收率与纯度。
9.根据本发明方法的另一变形形式,肟类中间体3的制备过程为:
樟脑和亚硝酸酯置于四氢呋喃中,在碱性条件下反应,反应完毕,经洗涤、中和、萃取、浓缩后得到肟化后的产物粗品,加水回流,反应完毕后萃取、浓缩,得到异构化肟类中间体3的粗品;其中,肟类中间体3的合成路线如下:。
10.需要说明的是,肟类中间体3的制备过程中,亚硝酸酯包含亚硝酸异戊酯;碱性条件所用碱包含氢氧化锂、氢氧化钠、强氧化钾或叔丁醇钾;反应温度为-45~r.t;回流时间为6~36h。
11.需要说明的是,肟类中间体3的制备过程中,樟脑、碱与亚硝酸酯的的摩尔比为1:1~2: 1~2。
12.根据本发明方法的另一变形形式,氨基酮类中间体4的制备过程为:将肟类中间体3粗品,置于-30~-15℃下缓慢滴加氢氧化钠溶液,降温等待温度稳定后,分批加入锌粉,然后自然升温使反应完毕,经萃取、浓缩得到氨基酮类中间体4粗品;其中,氨基酮类中间体4的合成路线如下:。
13.需要说明的是,氨基酮类中间体4的制备过程中,肟类中间体3和锌粉的摩尔比为1:1~5。
14.需要说明的是,氨基酮类中间体4的制备过程中,氢氧化钠溶液的浓度为1n至饱和溶液;反应温度为-45~r.t;具体优选为-30℃、-25℃、-20℃、-15℃、-10℃、-5℃、0℃、10℃、20℃。
15.根据本发明方法的另一变形形式,氨基醇类中间体5的制备过程为:将氢化铝锂溶解于乙醚中置于低温中冷却,等待温度稳定后,将氨基酮类中间体4粗品用乙醚溶解,随后慢慢滴加入反应瓶中,加完后静置,自然升温,待反应完毕后,加水淬灭后萃取、浓缩得到氨基醇类中间体5粗品;其中,氨基醇类中间体5的合成路线如下:。
16.需要说明的是,氨基醇类中间体5的制备过程中,反应温度为-45~r.t。
17.需要说明的是,氨基醇类中间体5的制备过程中,氨基酮类中间体4和氢化铝锂的摩尔比为1:1~5。
18.根据本发明方法的另一变形形式,氨基甲酸酯类中间体6的制备过程为:
将氨基醇类中间体5粗品置于dme中溶解,然后缓慢滴加氢氧化钠溶液,降温,滴加三光气的二氯甲烷溶液,加完后静置,自然升温,待反应完毕后,加水淬灭后萃取、浓缩得到粗品,通过柱色谱分离得到氨基甲酸酯类中间体6纯品;其中,氨基甲酸酯类中间体6的合成路线如下:。
19.需要说明的是,氨基甲酸酯类中间体6的制备过程中,氢氧化钠溶液的浓度为1n至饱和溶液;具体优选为2n、4n、6n、8n;反应温度为-45~r.t。
20.需要说明的是,氨基甲酸酯类中间体6的制备过程中,氨基醇类中间体5和三光气的摩尔比为1:0.3~1。
21.根据本发明方法的另一变形形式,格列波脲的制备过程为:将氨基甲酸酯类中间体6用乙醇和水溶解后,加入碱,加热回流,待反应完毕后,萃取、浓缩得到氨基醇7粗品,溶解在乙腈中并置于低温下,缓慢滴加tsnco,通过柱色谱分离得到格列波脲的纯品;格列波脲的合成路线如下:。
22.需要说明的是,格列波脲的制备过程中,氨基甲酸酯类中间体6和碱的摩尔比为1:1~8;氨基醇7和tsnco的摩尔比为1:0.5~2。
23.需要说明的是,格列波脲的制备过程中,碱包含氢氧化锂、氢氧化钠、强氧化钾、叔丁醇钾;具体优选为叔丁醇钾。
24.相对于现有技术,本发明的创新点:[1]本发明以易购的化工产品为原料,能够快速完成格列波脲的对映体的合成;[2]本发明的合成路线反应条件温和,易于调控,生产周期短,设计合理;[3] 本发明合成的格列波脲具有较高的收率与纯度;其hplc纯度大于99.5%。
[0025]
因此,本发明是一种能够得到收率与纯度较高的格列波脲。
具体实施方式
[0026]
下面结合具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0027]
需要说明的是,除非本发明清楚地要求,否则,在整个发明中,术语中“包含”、“含有”等被限定为一种包括含义,而不是排除性的或者穷举式的意思;也就是说,是“包括,但不限于”的意思。
[0028]
本发明提供了一种高纯格列波脲的制备方法,包括以下步骤:(1)肟类中间体3的制备
樟脑和亚硝酸酯置于四氢呋喃中,在碱性条件下反应,反应完毕,经洗涤、中和、萃取、浓缩后得到肟化后的产物粗品,加水回流,反应完毕后萃取、浓缩,得到异构化肟类中间体3的粗品;其中,肟类中间体3的合成路线如下:;(2)氨基酮类中间体4的制备将肟类中间体3粗品,置于-30~-15℃下缓慢滴加氢氧化钠溶液,降温等待温度稳定后,分批加入锌粉,然后自然升温使反应完毕,经萃取、浓缩得到氨基酮类中间体4粗品;其中,氨基酮类中间体4的合成路线如下:;(3)氨基醇类中间体5的制备将氢化铝锂溶解于乙醚中置于低温中冷却,等待温度稳定后,将氨基酮类中间体4粗品用乙醚溶解,随后慢慢滴加入反应瓶中,加完后静置,自然升温,待反应完毕后,加水淬灭后萃取、浓缩得到氨基醇类中间体5粗品;其中,氨基醇类中间体5的合成路线如下:;(4)氨基甲酸酯类中间体6的制备将氨基醇类中间体5粗品置于dme中溶解,然后缓慢滴加氢氧化钠溶液,降温,滴加三光气的二氯甲烷溶液,加完后静置,自然升温,待反应完毕后,加水淬灭后萃取、浓缩得到粗品,通过柱色谱分离得到氨基甲酸酯类中间体6纯品;其中,氨基甲酸酯类中间体6的合成路线如下:;(5)格列波脲的制备将氨基甲酸酯类中间体6用乙醇和水溶解后,加入碱,加热回流,待反应完毕后,萃取、浓缩得到氨基醇7粗品,溶解在乙腈中并置于低温下,缓慢滴加tsnco,通过柱色谱分离得到格列波脲的纯品;格列波脲的合成路线如下:
。
[0029]
需要说明的是,在本发明中,除非另有说明,缩写代表的含义如下表所示:以下结合具体实施方式对本发明的技术方案作进一步详细描述:实施例1:一种高纯格列波脲的制备方法,包括以下步骤:(1)肟类中间体3的制备具体反应路线如下:准确称取40.9 g樟脑和53.9g叔丁醇钾于反应瓶中,进行抽空换氮后,将反应瓶放置于-30℃低温下,在该温度下加入240ml四氢呋喃并搅拌均匀,然后缓慢滴加64.5ml亚硝酸异戊酯,滴加完继续搅拌1小时后,关掉制冷,让其自然升温,待反应完全后,加水萃取,留水相,对水相酸化后萃取,收集合并有机相,干燥,浓缩得固体,无需进一步纯化加水回流18小时,降至室温后萃取收集有机相,干燥,浓缩得肟类中间体3的粗品50 g;(2)氨基酮类中间体4的制备具体反应路线如下:将步骤(2)中的45 g肟类中间体3的粗品转移入反应瓶并放置于-15℃低温下,在这一温度下缓慢滴加300 ml 30%的氢氧化钠溶液。滴加完后,进一步降温到-30℃。待温度稳定后,分批加入58.7 g锌粉,加完后关掉制冷,让其自然升温,待反应完全后,萃取,收集合并有机相,干燥,浓缩得氨基酮类中间体4粗品38 g;
(3)氨基醇类中间体5的制备具体反应路线如下:取用干燥的反应瓶,投入27.3 g氢化铝锂和磁子,在低温下小心加入500 ml乙醚。另将上一步所得38 g氨基酮类中间体4粗品溶解于300 ml乙醚中配成溶液,缓慢滴加入反应瓶中。滴加完后继续搅拌1小时,关掉制冷,让其自然升温。待反应完全后,小心淬灭氢化铝锂,淬灭后萃取,收集合并有机相,干燥,浓缩得氨基醇类中间体5粗品38 g;(4)氨基甲酸酯类中间体6的制备具体反应路线如下:将步骤(3)所得38 g氨基醇类中间体5粗品溶解在1161 ml的dme中置于低温下-15℃低温下,在这一温度下滴加116 ml 6n的氢氧化钠溶液。滴加完后,进一步降温到-30℃。待温度稳定后,缓慢滴加三光气的二氯甲烷溶液(将23g三光气溶解在465ml二氯甲烷中)。滴加完后继续搅拌1小时,关掉制冷,让其自然升温。待反应完全后,加水,用乙酸乙酯萃取合并有机相用10%乙酸水溶液洗涤后,干燥,浓缩进行柱色谱分离(ea:pe = 2:3,rf ≈ 0.55),收集,浓缩得氨基甲酸酯类中间体6纯品9.5 g;(5)格列波脲的制备具体反应路线如下:取上步所得氨基甲酸酯类中间体6纯品9.5 g溶解在122 ml乙醇和65 ml水的混合溶液中搅拌均匀,加入7.8 g氢氧化钠,升温回流,待反应完全后,自然冷却到室温,萃取,干燥,浓缩得氨基醇中间体7 7 g;将所得到的氨基醇溶解在91 ml乙腈中置于0℃下,缓慢滴加6.6 ml 的tsnco,滴加完后继续搅拌1小时,关掉制冷,让其自然升温。待反应完全后,浓缩进行柱色谱分离(ea:dcm = 1:5,rf ≈ 0.3),收集,浓缩得格列波脲纯品11.2 g,产率73.9%, hplc纯度为99.58%。
[0030]1h nmr (400 mhz, dmso) δ 10.66 (s, 1h), 7.77 (d, j = 8.4 hz, 2h), 7.41 (d, j = 8.2 hz, 2h), 6.60 (d, j = 5.8 hz, 1h), 5.38 (d, j = 4.9 hz, 1h), 3.87-3.67 (m, 2h), 2.39 (s, 3h), 1.76-1.63 (m, 2h), 1.23 (d, j = 12.4 hz, 1h), 1.14-0.95 (m, 2h), 0.83 (d, j = 8.2 hz, 6h), 0.76 (s, 3h)。
[0031]
实施例2:一种高纯格列波脲的制备方法,与实施例1不同的是:(1)肟类中间体3的制备具体反应路线如下:准确称取69.8 g樟脑和51.5g叔丁醇钾于反应瓶中,进行抽空换氮后,将反应瓶放置于-30℃低温下,在该温度下加入240ml四氢呋喃并搅拌均匀,然后缓慢滴加64.5ml亚硝酸异戊酯,滴加完继续搅拌1小时后,关掉制冷,让其自然升温,待反应完全后,加水萃取,留水相,对水相酸化后萃取,收集合并有机相,干燥,浓缩得固体,无需进一步纯化加水回流18小时,降至室温后萃取收集有机相,干燥,浓缩得肟类中间体3的粗品80.62 g。
[0032]
实施例3:一种高纯格列波脲的制备方法,与实施例1不同的是:(2)氨基酮类中间体4的制备具体反应路线如下:将步骤(2)中的47.3g肟类中间体3的粗品转移入反应瓶并放置于-15℃低温下,在这一温度下缓慢滴加300 ml 30%的氢氧化钠溶液。滴加完后,进一步降温到-30℃。待温度稳定后,分批加入58.7 g锌粉,加完后关掉制冷,让其自然升温,待反应完全后,萃取,收集合并有机相,干燥,浓缩得氨基酮类中间体4粗品44.3 g。
[0033]
实施例4:一种高纯格列波脲的制备方法,与实施例1不同的是:(3)氨基醇类中间体5的制备具体反应路线如下:取用干燥的反应瓶,投入27.3 g氢化铝锂和磁子,在低温下小心加入500 ml乙醚。另将上一步所得24.0 g氨基酮类中间体4粗品溶解于300 ml乙醚中配成溶液,缓慢滴加入反应瓶中。滴加完后继续搅拌1小时,关掉制冷,让其自然升温。待反应完全后,小心淬灭氢化铝锂,淬灭后萃取,收集合并有机相,干燥,浓缩得氨基醇类中间体5粗品27.1 g。
[0034]
实施例5:
本发明在制备肟类中间体3过程中,将亚硝酸异戊酯替换为亚硝酸异戊酯与亚硝酸乙酯混合物,其中亚硝酸异戊酯与亚硝酸乙酯的摩尔比为1:1~2;将该混合物共同作为反应试剂,其能够提高肟类中间体3的产率,进而得到产率与纯度较高的格列波脲。
[0035]
具体地,一种高纯格列波脲的制备方法,与实施例1不同的是:步骤(1)肟类中间体3的制备中,将亚硝酸异戊酯替换为亚硝酸异戊酯与亚硝酸乙酯混合物,其中亚硝酸异戊酯与亚硝酸乙酯的摩尔比为1:1。
[0036]
实施例6:一种高纯格列波脲的制备方法,与实施例1不同的是:步骤(1)肟类中间体3的制备中,将亚硝酸异戊酯替换为亚硝酸异戊酯与亚硝酸乙酯混合物,其中亚硝酸异戊酯与亚硝酸乙酯的摩尔比为1:2。
[0037]
实施例7:一种高纯格列波脲的制备方法,与实施例1不同的是:步骤(1)肟类中间体3的制备中,将亚硝酸异戊酯替换为亚硝酸异戊酯与亚硝酸乙酯混合物,其中亚硝酸异戊酯与亚硝酸乙酯的摩尔比为1:0.5。
[0038]
实施例8:一种高纯格列波脲的制备方法,与实施例1不同的是:步骤(1)肟类中间体3的制备中,将亚硝酸异戊酯替换为亚硝酸异戊酯与亚硝酸乙酯混合物,其中亚硝酸异戊酯与亚硝酸乙酯的摩尔比为1:3。
[0039]
实施例9:为了进一步提高了格列波脲的收率与纯度,步骤(5)格列波脲的制备中,采用柱色谱分离进行分离时,将ea、dcm与二乙丙胺共同作为柱色谱柱的展开剂;其中ea、dcm与二乙丙胺=1:5:1~3。
[0040]
具体地,一种高纯格列波脲的制备方法,与实施例1不同的是:步骤(5)格列波脲的制备中,待反应完全后,浓缩进行柱色谱分离(ea:dcm :二乙丙胺= 1:5:1,rf ≈ 0.29),收集,浓缩得格列波脲纯品。
[0041]
实施例10:一种高纯格列波脲的制备方法,与实施例1不同的是:步骤(5)格列波脲的制备中,待反应完全后,浓缩进行柱色谱分离(ea:dcm :二乙丙胺= 1:5:3,rf ≈ 0.28),收集,浓缩得格列波脲纯品。
[0042]
试验例1:1. 肟类中间体3的收率本发明中实施例1、实施例2、实施例5-8中的肟类中间体3的收率如表1所示。
[0043]
表1 肟类中间体3的收率
由表1可以看出,实施例1与实施例2中肟类中间体3的收率不低于92%;即本技术能够得到收率较高的肟类中间体3;实施例5-6中肟类中间体3的收率高于96.5%,对比实施例1、实施例5-8,实施例5-6中肟类中间体3的收率高于实施例1、实施例7-8,这表明在制备肟类中间体3的过程中,将亚硝酸异戊酯与亚硝酸乙酯共同作为反应试剂,能够得到收率更高的肟类中间体3。
[0044]
2. 高纯格列波脲的收率与纯度采用高效液相色谱法对制得的高纯格列波脲的纯度进行测定。
[0045]
表2高纯格列波脲的收率与纯度由表2可以看出,实施例1中格列波脲的收率高于73%,纯度高于99.5%,实施例5-6中格列波脲的收率高于76%,纯度高于99.7%,对比实施例1、实施例5-8,实施例5-6中格列波脲的收率与纯度均高于实施例1、实施例7-8;这表明将亚硝酸异戊酯与亚硝酸乙酯共同作为反应试剂,得到收率较高的肟类中间体3,进而得到收率与纯度较高的格列波脲。实施例8-9中格列波脲的收率高于80%,纯度高于99.8%,对比实施例1、实施例8-9,实施例8-9中格列波脲的收率与纯度均高于实施例1,这表明在采用柱色谱分离进行分离时,将ea、dcm与二乙丙胺共同作为柱色谱柱的展开剂,其可能对格列波脲具有更好的极性选择性,进而得到收率与纯度的格列波脲。
[0046]
本发明的操作步骤中的常规操作为本领域技术人员所熟知,在此不进行赘述。
[0047]
以上所述的实施例对本发明的技术方案进行了详细说明,应理解的是以上所述仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充或类似方式替代等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。