1.本技术属于有机化学合成技术领域,具体涉及涉及一种2-(苯并噻吩-2
‑ꢀ
基)苯并[d]惡唑衍生物、制备方法及应用。
背景技术:
[0002]
2-(苯并噻吩-2-基)苯并[d]惡唑衍生物由于本身特殊的结构,使得其具有多方面的应用。在其结构的苯并噻吩环上的4,5,6,7位和苯并惡唑环上的 4,5,6,7位引入不同的活性基团,会在不同的领域都有不同的应用和更优异的性能,由于近年来科学家们对有机电至发光材料的不断研究和开发,2-(苯并噻吩-2-基)苯并[d]惡唑衍生物的苯并噻吩环上的4,5,6,7位和苯并惡唑环上的4,5,6,7位引入不同的活性基团需要越来越高的纯度和收率,而且随着oled有机发光材料的不断突破和市场化,对其中间体材料有了更大的需求,因此需要不停的创新和对工艺不断的进行优化和改进。
技术实现要素:
[0003]
有鉴于此,本发明的主要目的在于提供一种2-(苯并噻吩-2-基)苯并[d] 惡唑衍生物、制备方法及应用。
[0004]
为达到上述目的,本发明的技术方案是这样实现的:
[0005]
本发明实施例提供一种2-(苯并噻吩-2-基)苯并[d]惡唑衍生物,其特征在于,所述2-(苯并噻吩-2-基)苯并[d]惡唑衍生物通式为:
[0006][0007]
其中,r1为氢、烷基、卤素或者烷氧基,r2为氢、烷基、卤素或者烷氧基。
[0008]
上述方案中,所述烷基为c1~c40,优选为c1~c10。
[0009]
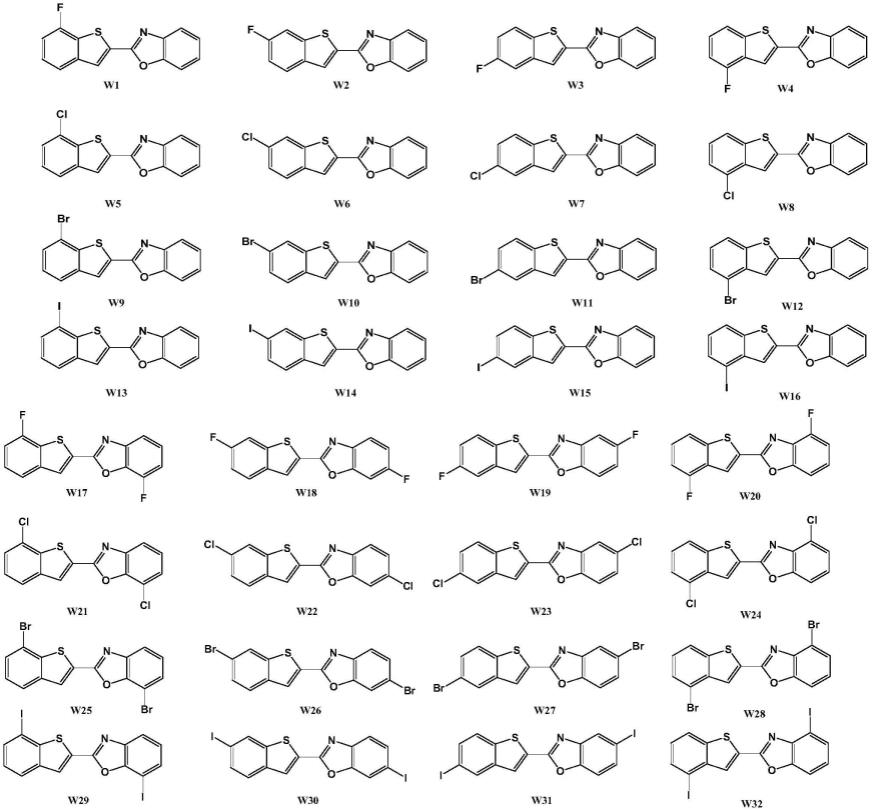
上述方案中,所述2-(苯并噻吩-2-基)苯并[d]惡唑衍生物选自下述具体结构式:
[0010]
[0011][0012]
本发明实施例还提供一种如上述方案中任意一项所述的2-(苯并噻吩-2
‑ꢀ
基)苯并[d]惡唑衍生物的其制备方法,该方法包括:
[0013]
以取代邻氟苯甲醛和巯基乙酸甲酯为原料先发生合环脱水得到第一中间体;
[0014]
所述第一中间体通过皂化得到第二中间体;
[0015]
所述第二中间体与取代邻氨基苯酚反应获得2-(苯并噻吩-2-基)苯并[d] 惡唑衍生物。
[0016]
上述方案中,所述以取代邻氟苯甲醛和巯基乙酸甲酯为原料先发生合环脱水得到第一中间体,具体为:在惰性气体保护下反应容器中加入溶剂,然后加入取代邻氟苯甲醛,再加入碱,最后滴加巯基乙酸甲酯,并且升温到60℃~ 120℃反应3~6小时,导入水中淬灭,通过乙酸乙酯萃取,浓缩后获得第一中间体。
[0017]
上述方案中,所述取代邻氟苯甲醛采用3-氯-2-氟苯甲醛、4-溴-2-氟苯甲醛、5-碘-2-氟苯甲醛或者2.6-二氟苯甲醛;所述溶剂采用二甲基亚砜、n,n
‑ꢀ
二甲基甲酰胺、nmp、二氧六环、二苯醚或者甲苯;所述碱采用碳酸钾、磷酸钾、三乙胺、吡啶、碳酸钠或者叔丁醇钠。
[0018]
上述方案中,所述第一中间体通过皂化得到第二中间体,具体为:将所述第一中间体溶于有机溶剂中并且加入碱,升温到30~80℃反应1~5小时,降温处理后获得第二中间
体。
[0019]
上述方案中,所述有机溶剂采用乙醇、甲醇、水或者异丙醇;所述碱采用氢氧化钠、氢氧化钾或者氢氧化锂;所述缩合剂采用ppa或者浓硫酸。
[0020]
上述方案中,所述第二中间体与取代邻氨基苯酚反应获得2-(苯并噻吩-2
‑ꢀ
基)苯并[d]惡唑衍生物,具体为:将所述第二中间体溶于缩合剂,加入磷酸,再加入取代邻氨基苯酚,所述取代邻氨基苯酚和第二中间体的摩尔比例为 1.5~0.8:1,升温到100℃~160℃,反应1~5小时后处理获得2-(苯并噻吩
ꢀ‑
2-基)苯并[d]惡唑衍生物。
[0021]
根据上述方案中任意一项制备的2-(苯并噻吩-2-基)苯并[d]惡唑衍生物的应用,将所述2-(苯并噻吩-2-基)苯并[d]惡唑衍生物应用合成药物、有机电至发光材料、高分子材料或者有机柔性材料。
[0022]
与现有技术相比,本发明的原材料都是市场上常规物料便宜易得且没有太多危险性,在简便的后处理之后能得到高纯度产品,产率高所以成本与其它方法比较更加的低廉,没有产生废溶剂,产生的废水少,原子利用率高所以更加的绿色环保。
附图说明
[0023]
此处所说明的附图用来公开对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0024]
图1为本发明实施例提供一种2-(苯并噻吩-2-基)苯并[d]惡唑衍生物的其制备方法的合成路线图。
具体实施方式
[0025]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0026]
需要说明的是,在本文中,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、物品或者装置不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、物品或者装置所固有的要素。在没有更多限制的情况下,由语句“包括一个
……”
限定的要素,并不排除在包括该要素的过程、物品或者装置中还存在另外的相同要素。
[0027]
本发明实施例提供一种2-(苯并噻吩-2-基)苯并[d]惡唑衍生物,所述 2-(苯并噻吩-2-基)苯并[d]惡唑衍生物通式为:
[0028][0029]
其中,r1为氢、烷基、卤素或者烷氧基,r2为氢、烷基、卤素或者烷氧基。
[0030]
所述烷基为c1~c40,优选为c1~c10。
[0031]
进一步地,所述2-(苯并噻吩-2-基)苯并[d]惡唑衍生物选自下述具体结构式:
[0032]
[0033][0034]
本发明实施例还提供一种2-(苯并噻吩-2-基)苯并[d]惡唑衍生物的其制备方法,如图1所示,该方法通过以下步骤实现:
[0035]
步骤101:以取代邻氟苯甲醛和巯基乙酸甲酯为原料先发生合环脱水得到第一中间体;
[0036]
具体地,在惰性气体保护下反应容器中加入溶剂,然后加入取代邻氟苯甲醛,再加入碱,最后滴加巯基乙酸甲酯,并且升温到60℃~120℃反应3~6小时,降温至50℃以下,导入水中淬灭,通过乙酸乙酯萃取,浓缩后获得第一中间体。
[0037]
所述取代邻氟苯甲醛采用3-氯-2-氟苯甲醛、4-溴-2-氟苯甲醛、5-碘-2
‑ꢀ
氟苯甲醛或者2.6-二氟苯甲醛等;
[0038]
所述溶剂采用二甲基亚砜、n,n-二甲基甲酰胺、nmp、二氧六环、二苯醚或者甲苯等;
[0039]
所述碱采用碳酸钾、磷酸钾、三乙胺、吡啶、碳酸钠或者叔丁醇钠等。
[0040]
惰性气体为氮气或者氩气。
[0041]
所述取代邻氟苯甲醛和巯基乙酸甲酯摩尔比例为1:0.8~1.5;优选的,所用取代邻氟苯甲醛和巯基乙酸甲酯摩尔比例为1:1,
[0042]
步骤102:所述第一中间体通过皂化得到第二中间体;
[0043]
具体地,将所述第一中间体溶于有机溶剂中并且加入碱,升温到30~80℃反应1~5小时,降温处理后获得第二中间体。
[0044]
所述有机溶剂采用乙醇、甲醇、水或者异丙醇;所述碱采用氢氧化钠、氢氧化钾或者氢氧化锂。
[0045]
步骤103:所述第二中间体与取代邻氨基苯酚反应获得2-(苯并噻吩-2
‑ꢀ
基)苯并[d]惡唑衍生物。
[0046]
具体地,将所述第二中间体溶于缩合剂,加入磷酸,再加入取代邻氨基苯酚,所述取代邻氨基苯酚和第二中间体的摩尔比例为1.5~0.8:1,升温到 100℃~160℃,反应1~5小时后处理获得2-(苯并噻吩-2-基)苯并[d]惡唑衍生物。
[0047]
所述缩合剂采用ppa或者浓硫酸。
[0048]
本发明的制备方法,原材料都是市场上常规物料便宜易得且没有太多危险性,第一步反应采用的是取代脱水缩合反应,采用常规反应釜即可,合成操作过程简便容易操作,相对于其它方案本方案设计的反应步骤更少只有三步反应,相应的能耗更少,后处理更加简单。
[0049]
本发明的制备方法,在简便的后处理之后能得到高纯度产品,产率高所以成本与其它方法比较更加的低廉,没有产生废溶剂,产生的废水少,原子利用率高所以更加的绿色环保。
[0050]
实施例1:
[0051][0052]
向1000ml反应器中加入,2.5-二氟苯甲醛(30.0g,0.21mol),溶于300ml 二甲基亚砜中,再加入三乙胺(64.1g,0.63mol),然后滴加巯基乙酸甲酯 (24.6g,0.23mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s1,5-氟苯并噻吩-2-甲酸甲酯,43.9g,hplc=96.5,收率 99.0%。
[0053]
向2000ml反应器中加入5-氟苯并噻吩-2-甲酸甲酯(20g,0.10mol),溶于 800ml乙醇,再加入200ml水中,加入氢氧化钾(16.0g,0.29mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s2,5-氟苯并噻吩
ꢀ‑
2-甲酸,18.3g,hplc=98.5,收率98.1%。
[0054]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入5-氟苯并噻吩-2-甲酸(15.0g,0.08mol),邻氨基苯酚 (10.0g,0.09mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w3,2-(5-氟苯并噻吩-2-基)苯并惡唑,17.5g,hplc=99.1,收率85%。
[0055]
实施例2:
[0056][0057]
向1000ml反应器中加入,4-溴-2-氟苯甲醛(30.0g,0.15mol),溶于 300mln,n-二甲基甲酰胺中,再加入碳酸钾(61.2g,0.44mol),然后滴加巯基乙酸甲酯(15.7g,0.15mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s3,6-溴苯并噻吩-2-甲酸甲酯,38.8g,hplc=96.8,收率97.0%。
[0058]
向2000ml反应器中加入6-溴苯并噻吩-2-甲酸甲酯(20g,0.07mol),溶于 800ml甲醇,再加入200ml水中,加入氢氧化钠(8.9g,0.22mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s4,6-溴苯并噻吩
ꢀ‑
2-甲酸,18.1g,hplc=97.9,收率95.5%。
[0059]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入6-溴苯并噻吩-2-甲酸(15.0g,0.08mol),邻氨基苯酚 (13.1g,0.12mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w10,2-(6-溴苯并噻吩-2-基)苯并惡唑,20.3g,hplc=99.5,收率77%。
[0060]
实施例3:
[0061][0062]
向1000ml反应器中加入,4-氯-2-氟苯甲醛(30.0g,0.19mol),溶于 300mlnmp,再加入磷酸钾(120g,0.57mol),然后滴加巯基乙酸甲酯 (15.7g,0.15mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s5,6-氯苯并噻吩-2-甲酸甲酯,39.6g,hplc=94.9,收率 92.8%。
[0063]
向2000ml反应器中加入6-氯苯并噻吩-2-甲酸甲酯(20g,0.09mol),溶于 800ml叔丁醇,再加入200ml水中,加入氢氧化钾(14.8g,0.26mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙
酯重结晶后得到中间体s6,6-氯苯并噻吩
ꢀ‑
2-甲酸,18.1g,hplc=96.8,收率96.2%。
[0064]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入6-氯苯并噻吩-2-甲酸(15.0g,0.07mol),5-氯-2-氨基苯酚(10.1g,0.07mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w22,6-氯
‑ꢀ
2-(6
’‑
溴苯并噻吩-2
’‑
基)苯并惡唑,15.5g,hplc=98.9,收率69%。
[0065]
实施例4:
[0066][0067]
向1000ml反应器中加入,3-溴-2-氟苯甲醛(30.0g,0.15mol),溶于 300mln,n-二甲基甲酰胺中,再加入碳酸钠(47.7g,0.45mol),然后滴加巯基乙酸甲酯(23.3g,0.22mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s7,7-溴苯并噻吩-2-甲酸甲酯,38.4g,hplc=97.2,收率94.5%。
[0068]
向2000ml反应器中加入7-溴苯并噻吩-2-甲酸甲酯(20g,0.07mol),溶于 800ml乙醇,再加入200ml水中,加入氢氧化钾(11.8g,0.21mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s8,7-溴苯并噻吩
ꢀ‑
2-甲酸,17.6g,hplc=96.5,收率98.1%。
[0069]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入7-溴苯并噻吩-2-甲酸(15.0g,0.08mol),6-氟-2-氨基苯酚(12.2g,0.10mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w33,7-氟
ꢀ‑
2-(7
’‑
溴苯并噻吩-2
’‑
基)苯并惡唑,22.0g,hplc=99.0,收率79%。
[0070]
实施例5:
[0071]
[0072]
向1000ml反应器中加入,3-溴-2-氟苯甲醛(30.0g,0.15mol),溶于300ml 二甲基亚砜中,再加入吡啶(33.3g,0.45mol),然后滴加巯基乙酸甲酯(15.9g,0.15mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s9,5-溴苯并噻吩-2-甲酸甲酯,39.7g,hplc=96.2,收率 97.7%。
[0073]
向2000ml反应器中加入5-溴苯并噻吩-2-甲酸甲酯(20g,0.07mol),溶于 800ml乙醇,再加入200ml水中,加入氢氧化钠(8.4g,0.21mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s10,5-溴苯并噻吩-2-甲酸,16.9g,hplc=96.5,收率94.2%。
[0074]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入5-溴苯并噻吩-2-甲酸(15.0g,0.08mol),4-甲氧基-2
‑ꢀ
氨基苯酚(10.0g,0.08mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w33,5
‑ꢀ
甲氧基-2-(5
’‑
溴苯并噻吩-2
’‑
基)苯并惡唑,19.9g,hplc=98.5,收率72%。
[0075]
实施例6:
[0076][0077]
向1000ml反应器中加入,3-氯-2-氟苯甲醛(30.0g,0.19mol),溶于 300mln,n-二甲基亚砜,再加入三乙胺(57.6g,0.57mol),然后滴加巯基乙酸甲酯(26.2g,0.25mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s11,7-氯苯并噻吩-2-甲酸甲酯,41.3g,hplc=95.3,收率 95.8%。
[0078]
向2000ml反应器中加入7-氯苯并噻吩-2-甲酸甲酯(20g,0.09mol),溶于 800ml甲醇,再加入200ml水中,加入氢氧化钠(10.8g,0.27mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s12,7-氯苯并噻吩
ꢀ‑
2-甲酸,33.2g,hplc=97.2,收率95.9%。
[0079]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入7-氯苯并噻吩-2-甲酸(15.0g,0.07mol),6-甲基-2-氨基苯酚(11.2g,0.09mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w53,7-甲基
‑ꢀ
2-(7
’‑
氯苯并噻吩-2
’‑
基)苯并惡唑,14.9g,hplc=98.3,收率71%。
[0080]
实施例7:
[0081][0082]
向1000ml反应器中加入,5-氯-2-氟苯甲醛(30.0g,0.19mol),溶于 300mlnmp,再加入磷酸钾(120g,0.57mol),然后滴加巯基乙酸甲酯 (20.1g,0.19mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s13,5-氯苯并噻吩-2-甲酸甲酯,40.2g,hplc=94.9,收率 93.3%。
[0083]
向2000ml反应器中加入5-氯苯并噻吩-2-甲酸甲酯(20g,0.09mol),溶于 800ml乙醇醇,再加入200ml水中,加入氢氧化钾(14.8g,0.26mol),在20-50℃,反应1-8小时,溶液变浑浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s14,5-氯苯并噻吩-2-甲酸,33.6g,hplc=97.5,收率96.9%。
[0084]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入5-氯苯并噻吩-2-甲酸(15.0g,0.07mol),4-溴-2-氨基苯酚(14.5g,0.08mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w63,5-溴
‑ꢀ
2-(5
’‑
溴苯并噻吩-2
’‑
基)苯并惡唑,18.4g,hplc=98.8,收率72%。
[0085]
实施例8:
[0086][0087]
向1000ml反应器中加入,3-溴-2-氟苯甲醛(30.0g,0.15mol),溶于 300mlnmp中,再加入碳酸钾(62.1g,0.45mol),然后滴加巯基乙酸甲酯(22.3g,0.21mol),氮气保护下,升温到60℃-120℃,加热反应1-5小时,反应结束冷却至室温,使用乙酸乙酯和水萃取,有机相干燥后抽滤,得到滤液减压浓缩,得中间体s15,7-溴苯并噻吩-2-甲酸甲酯,39.0g,hplc=96.0,收率 96.0%。
[0088]
向2000ml反应器中加入7-溴苯并噻吩-2-甲酸甲酯(20g,0.07mol),溶于 800ml乙醇,再加入200ml水中,加入氢氧化钾(11.8g,0.21mol),在20-50℃,反应1-8小时,溶液变浑
浊,监控反应结束,调pe至4左右,用乙酸乙酯萃取,减压浓缩得到白色固体,再用乙酸乙酯重结晶后得到中间体s16,7-溴苯并噻吩-2-甲酸,16.8g,hplc=96.1,收率94.0%。
[0089]
向1000ml反应器中加入多聚磷酸120ml,磷酸80ml,升温到100℃-130℃,开启搅拌,然后加入7-溴苯并噻吩-2-甲酸(15.0g,0.08mol),4-溴-2-氨基苯酚(16.5g,0.09mol),升温到130℃-170℃,保温反应1-8小时,降温后,导入水中淬灭,抽滤得到粗品,然后用甲苯重结晶得到合格固体w65,5-溴
ꢀ‑
2-(7
’‑
溴苯并噻吩-2
’‑
基)苯并惡唑,24.9g,hplc=99.0,收率76%。
[0090]
本发明实施例还提供一种2-(苯并噻吩-2-基)苯并[d]惡唑衍生物的应用,将所述2-(苯并噻吩-2-基)苯并[d]惡唑衍生物应用合成药物、有机电至发光材料、高分子材料或者有机柔性材料。
[0091]
以上所述,仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。