1.本发明涉及免疫治疗技术领域,具体涉及一种可诱导嵌合抗原受体及其应用。
背景技术:
2.全球老龄化社会导致阿尔兹海默病(ad)等老年期痴呆在2016年已超过5000万,直接医疗费用及间接照料支出巨大。而ad已有药物只能对症缓解,并不能延缓病程,近16年来3000多种药物均告失败。提示仅从ad的病理学特征-β淀粉样蛋白和过磷酸化tau进行“头痛医头”式单一靶点治疗很难成功,而应从慢性炎症等促进疾病进展等病理机制进行多靶点干预治疗新策略探索。美国fda宣布ad新药研发的重大变化:与既往新药研发集中在出现明显认知障碍的3期和4期阶段相比,将支持极早期(1期:出现ad病理变化,但没有任何临床症状;2期:病理变化基础上出现微小且可测的神经生理学异常,也没有出现功能上的衰退)的疾病干预,明确提出采用生物标志物作为新的临床终点。
3.嵌合抗原受体t细胞(chimeric antigen receptormodified t cells,car-t)是由抗原特异性的受体或特异性识别抗原的单克隆抗体的scfv、胞外间隔序列、细胞跨膜区和胞内t细胞活化结构域串联而成。作为特异性细胞免疫疗法之一,car-t细胞在肿瘤领域进展日新月异。m1型小胶质细胞因子是引发神经系统炎症的主要细胞,对ad的发生发展具有明确的促进作用,csf1r在m1型小胶质细胞表面高表达,是理想的靶向治疗靶点。因此,靶向csf1r的car-t细胞治疗ad在理论上可行。然而,由于car-t细胞具有非常强的靶向杀伤作用,可以在短时间快速杀伤大量靶细胞后,释放大量的细胞因子,从而诱发“细胞因子风暴”(cytokine storm,crs)。因此,控制csr的副作用在临床治疗中尤其重要。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种嵌合抗原受体,通过利用可诱导表达系统,实现对car基因表达水平进行调控,避免了car-t细胞发挥治疗作用时可能触发的“细胞因子风暴”副作用。
5.为解决上述技术问题,本发明提供了以下技术方案:
6.本发明提供了一种嵌合抗原受体,所述嵌合抗原受体包括:抗人csf1r的单链抗体scfv、截短的higg1铰链区sh、t细胞共刺激信号分子以及t细胞胞内信号结构域;所述嵌合抗原受体的转录由可诱导表达元件调控,所述可诱导表达元件为tet operater。
7.优选的,所述tetoperater的核苷酸序列如seq id no.5所示。
8.优选的,所述截短的higg1铰链区sh的氨基酸序列如seq id no.4所示。
9.优选的,所述t细胞共刺激信号分子包括cd28、41bb、ox40中的一种或多种。
10.优选的,所述t细胞胞内信号结构域为cd3 zeta胞内信号活化结构域。
11.优选的,所述嵌合抗原受体的氨基末端连接信号肽,所述信号肽的氨基酸序列如seq id no.3所示。
12.优选的,所述嵌合抗原受体的核苷酸序列如seq id no.1所示,所述嵌合抗原受体
的氨基酸序列如seq id no.2所示。
13.本发明提供了一种重组慢病毒,所述重组慢病毒包含上述的嵌合抗原受体。
14.本发明还提供了一种car-t细胞,所述car-t细胞表达上述的嵌合抗原受体;或所述car-t细胞包括上述的重组慢病毒。
15.本发明还提供了上述的嵌合抗原受体、重组慢病毒或car-t细胞在制备治疗阿尔茨海默病药物中的应用。
16.本发明提供了一种嵌合抗原受体,可以利用病毒载体制备得到包含所述嵌合抗原受体的慢病毒假型病毒颗粒,利用制备的慢病毒转导活化t细胞,制备成可诱导的car-t细胞。本发明嵌合抗原受体表达时间与表达水平通过诱导剂dox添加时间和剂量进行调控,在dox诱导剂存在情况下,car-t细胞特异性杀伤csf1r阳性的靶细胞,分泌细胞因子il-2和ifn-γ;在撤掉诱导剂dox后,car-t细胞几乎对csf1r阳性细胞不产生杀伤作用,不再分泌细胞因子il-2和ifn-γ。经实验证实,采用本发明的car-t细胞能调控性杀伤靶细胞,提高了对ad治疗的靶向性;同时,本发明通过利用可诱导表达系统,实现了对car基因表达水平的调控,能够控制细胞因子分泌水平,有效避免了car-t细胞发挥治疗作用可能触发的“细胞因子风暴”副作用。
附图说明
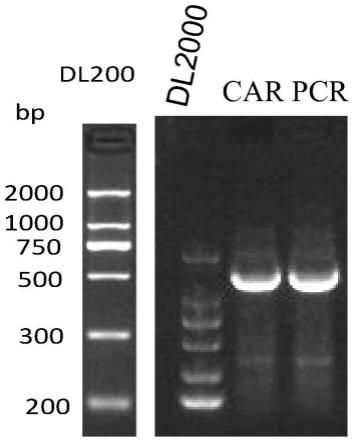
17.图1为本发明所述的合成csr1 car基因利用pcr扩增后dna片段。
18.图2为本发明所述的载体质粒dna双酶切。
19.图3为本发明所述的合成car基因亚克隆到慢病毒可诱导载体plvx-teton上菌落pcr验证结果。
20.图4为本发明所述的合成car基因亚克隆到慢病毒可诱导载体plvx-teton上酶切验证结果。
21.图5为加dox诱导够48h,利用流式细胞术检测csf1 car慢病毒转导在t细胞中的表达。
22.图6为加dox诱导够48h,利用qpcr检测csf1 car mrna表达水平。
23.图7为facs检测a549-csf1r-gfp稳转细胞株的csf1r表达率。
24.图8为mts检测本发明中csf1 car-t细胞在dox诱导前后对靶细胞后a549-csf1r杀伤作用。
25.图9为本发明中可诱导csf1r.car-t杀伤靶细胞后il-2表达。
26.图10为本发明中可诱导csf1r.car-t杀伤靶细胞后ifn-γ表达。
具体实施方式
27.本发明提供了一种嵌合抗原受体,所述嵌合抗原受体包括:抗人csf1r的单链抗体scfv、截短的higg1铰链区sh、t细胞共刺激信号分子以及t细胞胞内信号结构域;所述嵌合抗原受体的转录由可诱导表达元件调控,所述可诱导表达元件为tet operater。
28.本发明中,所述tet operater含tta结合原件,所述tet operater的核苷酸序列优选如seq id no.5所示。本发明所述tetoperater可诱导调控car的表达。本发明中,所述抗人csf1r的单链抗体scfv优选的来源鼠抗人csf1r蛋白的单克隆抗体,所述单链抗体scfv的
氨基酸序列如seq id no.7所示。本发明中,所述截短的higg1铰链区sh的氨基酸序列优选为pro lys ser cysasp lys thrhis thr cys pro pro cys(seq id no.4)。本发明所述截短的higg1铰链区可以缩小car-t细胞鱼肿瘤细胞的距离从而赋予car-t细胞具有更强的肿瘤细胞杀伤能力。
29.本发明中,所述t细胞共刺激信号分子优选包括cd28、41bb、ox40中的一种或多种。所述t细胞胞内信号结构域优选为cd3 zeta胞内信号活化结构域。本发明中,所述t细胞共刺激信号分子和t细胞胞内信号结构域优选的串联作为胞内活化结构域;所述胞内活化结构域优选包括cd28-ox40-cd3zeta,cd28-cd3zeta,41bb-cd3zeta,cd28-41bb-cd3zeta。
30.本发明中,所述嵌合抗原受体的氨基末端优选的连接信号肽,所述信号肽的氨基酸序列优选如seq id no.3所示。本发明中,所述信号肽优选的在car表达后被切割掉,所述信号肽可引导蛋白定位于细胞膜上。
31.本发明中,所述嵌合抗原受体的核苷酸序列优选如seq id no.1所示,所述嵌合抗原受体的氨基酸序列优选如seq id no.2所示。本发明所述嵌合抗原受体的重链和轻链分子之间的连接肽序列优选为(gly4ser)3,具体序列优选为:gly gly gly gly ser gly gly gly gly ser gly gly gly gly ser(seq id no.6)。
32.本发明提供了一种重组慢病毒,所述重组慢病毒包含上述的嵌合抗原受体。本发明中,所述scfv序列经人源密码子优化后,在5’端添加信号肽序列,3’端添加sh-胞内活化结构域,全基因合成后克隆入可诱导慢病毒骨架载体,即获得重组慢病毒。本发明对所述慢病毒骨架载体并没有特殊限定,采用本领域常规慢病毒骨架载体即可。
33.本发明还提供了一种car-t细胞,所述car-t细胞表达上述的嵌合抗原受体;或所述car-t细胞包括上述的重组慢病毒。本发明中,所述car-t细胞优选的由上述重组慢病毒与辅助质粒共同转导293t细胞生产得到自我灭活的慢病毒假颗粒后,转导活化t细胞后所得。本发明中,所述辅助质粒优选的包括pmd2g、pspax2。本发明所述car-t细胞可经诱导剂诱导表达,所述诱导剂优选为多西环素dox。在所述多西环素dox存在情况下,car-t细胞特异性杀伤csf1r阳性的靶细胞,分泌细胞因子il-2和ifn-γ;在撤掉诱导剂dox后,car-t细胞几乎对csf1r阳性细胞不产生杀伤作用,不再分泌细胞因子il-2和ifn-γ。
34.本发明还提供了上述的嵌合抗原受体、重组慢病毒或car-t细胞在制备治疗阿尔茨海默病药物中的应用。本发明分别在有或无诱导剂dox存在情况检测car的表达并检测其对csf1r阳性细胞的杀伤作用和细胞因子分泌水平,从而使所述的car-t细胞能清除csf1r阳性的m1型小胶质细胞,可以用于ad靶向治疗的药物。
35.为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例对本发明进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
36.下述实施例中,如无特殊说明,均为常规方法。
37.下述实施例中所用的材料、试剂、培养皿等,如无特殊说明,均可从商业途径得到。
38.实施例1可诱导csf1r.car慢病毒载体克隆构建
39.1.pcr扩增csf1r car dna片段
40.如序列1所示,拼接的dna序列由南京金斯瑞生物技术有限公司进行合成,合成的csf1r car克隆在puc57载体中,命名为puc57-csf1rcar。以该质粒为模板,利用上下游引物扩增csf1rcar dna片段。其中,上下游引物的序列如下:
41.上游引物为:5'-gaggtggtctggatccttagcgagggggcagggcctgcatgtga-3'(seq id no.8);
42.下游引物为:5'-ccctcgtaaagaattcatggccctgcccgtgaccgctctgct-3'(seq id no.8)。
43.扩增反应体系为:2
×
primerstarbuffer(购至takara公司)25μl,上下游引物分别3μl,模板puc57-csf1rcar质粒dna 5ng,用去离子灭菌水(ddw)补到50μl。
44.pcr扩增条件为:98℃10sec,60℃10sec,72℃10sec,扩增30个循环,72℃5min。
45.pcr扩增产物用琼脂糖凝胶电泳分离,结果如图1。从琼脂糖凝胶电泳结果可以看出,pcr产物大小约1500bp,与预期大小相符。
46.2.载体线性化
47.制备plvx-tetone-sv40p-puro质粒dna,用限制性内切酶ecori和bamhi(neb公司)进行双酶切,酶切体系为:质粒dna 2μg,10
×
cutsartbuffer 2μl,ecor i和bamh i各1μl,用去离子灭菌水(ddw)补到20μl,酶切反应体系在37℃水浴孵育4h。
48.酶切后的产物经琼脂糖凝胶电泳分离后,用琼脂糖凝胶dna回收试剂盒(购置捷瑞生物工程有限公司)回收载体dna,结果如图2。从琼脂糖凝胶电泳结果可以看出,载体dna内完全线性化。
49.3.插入片段和载体分子连接、转化
50.将琼脂糖凝胶凝胶电泳分离的pcr产物与载体片段按照分子摩尔比4:1混合,然后加入2μl 5
×
无缝克隆链接混合物(购于镇江爱必梦生物科技有效公司),最后用ddw补至10μl,链接混合物置于冰上30min,然后转化stble3感受态细菌(购于镇江维根生物科技有限公司)。细菌转化后涂布于含有氨卞青霉素的lb琼脂培养皿,37℃培养过夜。
51.4.阳性重组克隆筛选鉴定
52.分别挑取10个过夜培养的单菌落于20μl ddw中,充分混匀,开水煮沸5min,13000rmp离心5min,吸取2μl上清作为模板,进行pcr扩增筛选阳性克隆,pcr扩增反应体系和条件如步骤1所述。
53.pcr扩增产物经琼脂糖凝胶电泳筛选阳性克隆,结果如图3所示。3、5、7、9为阳性克隆扩增后抽提质粒dna,然后用ecori和xhoi双酶切验证,结果如图4所示。可以看出,双酶产生720bp的dna片段,与预期结果相符合。阳性克隆进一步送苏州金唯智生物科技有限公司测序验证。
54.5.高纯度去内毒素质粒制备:按照omega(购于omega公司)去内毒素质粒制备试剂盒的说明书制备高纯度的去内毒素质粒,具体步骤如下:
55.1)在15ml玻璃试管中加入4ml lb培养液,再加入4μl相应抗生素,平板上挑选单菌落加入长玻璃试管中,放置37℃200rpm振荡培养5.5小时;
56.2)将培养的带细菌液体倒入含有30ml lb培养基的1l烧瓶中,并加入35μl氨苄青霉素(1:1000),放置37℃,200rpm振荡培养至次日;
57.3)将菌液倾倒于50ml离心管,3500rmp,离心15min收集管底细菌;
58.4)用1ml solution 1/rnasea(保存于4℃冰箱)完全重悬细菌,反复吹吸,然后振荡器上震荡30-60s,将菌液均分到2个2ml离心管(即500ul/管);
59.5)每管加入500μl solution ii(solution ii若有沉淀,需加热),轻柔翻转10~
15次,温室静置3min;
60.6)每管加入250μl n3上下颠倒混匀10次,静置3min;13000rmp,15min高速离心;将上清转移到一新离心管(沉淀丢弃),并测量上清体积;
61.7)在上清中加入0.1倍体积的etr solution(保存于4℃冰箱);上下彻底混匀;(如果转移的上清是1200μl,请加入120μl etr solution)(应该是triton x-114);
62.8)将离心管插入冰中静置10min,每2~3min拿出上下颠倒几次(加入etr solution后,裂解液将变混浊,冰浴后将变澄清);
63.9)冰浴10min后将裂解液置于42℃水浴5min;裂解液将再次变浑浊(备注:将水浴温度调至70℃用于温浴e.b);
64.10)25℃,13000rmp离心8min;etr solution沉淀于离心管底部(沉淀为多糖、内毒素);将上清转移至2ml新离心管;加0.5倍无水乙醇;上下颠倒10次后室温放置2min;
65.11)将2个平衡过的hibind dna收集柱ii插入2ml收集管;加入700μl细菌裂解液上清;13000rmp离心1min后弃滤过液;
66.12)在hibind dna收集柱ii再次加入剩余细菌裂解液;13000rmp离心1min后弃滤过液;如此反复直至所有细菌裂解液全部被过滤;
67.13)在hibind dna收集柱ii中加入500μl hbc buffer(新hbc使用前必须加入异丙醇);13000rmp离心1min后弃滤过液;
68.14)在hibind dna收集柱ii中加入700μl dna wash buffer(新dna wash buffer使用前必须加入100%乙醇);13000rmp离心1min后弃滤过液;
69.15)700μl dna wash buffer再洗一次dna收集柱;丢弃滤过液后再次13000rmp离心2min,以确保完全去除dna收集柱中残留的乙醇;
70.16)将dna收集柱置于新的1.5ml无菌离心管(剪去管盖以便离心),室温再静置5min让乙醇完全挥发干净;在膜中央加120μl70℃预热的elution buffer(洗脱液);室温放置1min;
71.17)13000rmp离心1min;在超净工作台将两管洗脱液合并于一新1.5ml无菌离心管,然后测dna浓度并将dna保存于-80℃冰箱。
72.实施例2可诱导csf1r.car修饰的t淋巴细胞制备
73.1.可诱导csf1r.car慢病毒包装
74.1.1 293t细胞接种
75.将生长状态良好的293t细胞用0.25%胰蛋白酶消化制备单细胞悬液并调整浓度为4
×
105/ml,取10ml接种于10cm培养皿中,37℃,5%co2培养箱内培养过夜,第二天细胞汇合度达到80%。
76.1.2质粒转染
77.将3μg pmd2g、6μg pspax2和car慢病毒载体质粒7.5μg加到150μl opti-mem培养基内混匀;取25μl lipo2000加入到500μlopti-mem培养基内混匀,室温静置5min,将质粒混合物缓慢加入lipo 2000,混匀后室温静置15min。逐滴加入到培养皿内,充分混匀。6h后更换为含10%fbs的dmem新鲜培养液。
78.1.3收集上清并浓缩病毒
79.上述细胞培养48h后收集含慢病毒的培养基上清,1500g,4℃,离心10min去细胞碎
片。然后用0.45μm滤膜过滤病毒上清液。过滤液按照4:1的比例加入5
×
pegit病毒浓缩液。4℃静置过夜后,3200g,4℃,离心10min。弃去上清,用含5%fbs的dmem培养液重悬病毒团块。200μl/管分装,-80℃保存。纯化后的病毒通过qpcr滴度测定试剂盒(购自镇江爱必梦生物科技有限公司)测定病毒滴度,测定所得的病毒滴度为1
×
108pfu/ml。
80.2.pbmc分离、活化
81.2.1 pbmc分离
82.取健康血浆捐赠人的外周静脉血10ml于抗凝管中。等体积pbs稀释后利用淋巴细胞分离液进行密度梯度离心获取pbmc细胞。
83.成功分离pbmc细胞后,将其接种于包含有10%fbs的t551培养基中于37℃/5%co2细胞培养箱中培养。
84.2.2 pbmc活化
85.利用包含有1μl/ml il-2的t细胞培养基制备1
×
106/ml的pbmc细胞悬液。将细胞接种于利用cd3/cd28抗体包被的24孔细胞培养板获取活化t细胞,具体活化参数如下表1。
86.表1活化条件
[0087][0088]
3.car-t细胞制备
[0089]
利用步骤1所制备慢病毒按照moi=20转导步骤2活化的t细胞,制备可诱导csf1r.car修饰的t淋巴细胞。
[0090]
4.利用dox对csf1r分子表达进行诱导
[0091]
将步骤3制备好的car-t细胞培养于24孔板中,添加1μg/ml的dox诱导t细胞表达csf1r.car分子。
[0092]
5.流式细胞术检测csf1r.car分子表达
[0093]
利用可以识别car中fab片段的流式细胞检测抗体对已经制备的car-t细胞进行csf1r.car表达效率的检测,即取2
×
105的诱导后car-t细胞用anti-mfab-apc抗体进行染色30min,然后通过流式细胞仪分析car表达阳性率,结果如图5所示。
[0094]
可以看出,本发明嵌合抗原受体表达时间与表达水平通过诱导剂dox添加时间和剂量进行调控,car-t细胞经dox诱导后car表达率提高。
[0095]
实施例3 csf1r.car-t细胞对csf1r过表达靶细胞的杀伤作用检测
[0096]
1.mts法检测杀伤作用
[0097]
将靶细胞与诱导后car-t细胞按照不同效靶比于96孔板中进行共培养后,取csf1r过表达的靶细胞,并利用流式细胞分析确定其csf1r的表达效率,结果如图7所示,表明a549细胞的csf1r阳性率高达94%。
[0098]
在t细胞与a549-csf1r-gfp靶细胞胞共培养24h后,利用mts法检测杀伤后剩余肿瘤细胞的数量所确定的t细胞杀伤效果,结果如图8所示,表明本发明可诱导car-t细胞经dox诱导后,对靶细胞具有显著的杀伤作用,而未转导的活化t细胞与未经dox诱导的对照car-t细胞则无显著杀伤作用。
[0099]
2.elisa法检测细胞因子表达
[0100]
为验证csf1r.car-t细胞杀伤靶细胞后活化情况,将t细胞与靶细胞细胞共培养并24h和48h后收取上清,利用elisa检测上清中il-2和ifn-γ的含量。elisa实验步骤参照elisa试剂盒(购自美国bd公司)进行。
[0101]
具体操作步骤如下:
[0102]
(1)抗体包被:按照试剂盒说明书将包被抗体稀释后加入elisa专用96孔板,每孔100μl,4℃冰箱孵育过夜;
[0103]
(2)收集上清:收集共培养杀伤实验的培养基上清并作相应稀释;
[0104]
(3)封闭:弃去包被液,加入洗液,重复清洗3次;每孔加入200μl 1
×
assay buffer,室温孵育1h封闭包被孔;
[0105]
(4)添加样品和标准品:弃封闭液,加入洗液,洗3次;每孔加入100μl系列浓度的标准品或样品,室温孵育2h;
[0106]
(5)洗涤:弃去样品液和标准品液,加入洗液,清洗5次;
[0107]
(6)添加检测抗体和辣根过氧化物酶:每孔加入100μl稀释后检测抗体和辣根过氧化物酶混合液,室温孵育1h;然后加入洗液,洗7次;
[0108]
(7)显色:每孔加入100μl tmb底物液,37℃,避光孵育5-30min;每孔加入50μl终止液终止反应;30min内,在酶标仪450nm波长下读板;
[0109]
(8)根据标准品的od450值建立标准曲线,并计算样品细胞因子浓度,得到结果图9和图10。
[0110]
由上述实施例可以看出,本发明嵌合抗原受体表达时间与表达水平通过诱导剂dox添加时间和剂量进行调控,在dox诱导剂存在情况下,car-t细胞特异性杀伤csf1r阳性的靶细胞,分泌细胞因子il-2和ifn-γ;在撤掉诱导剂dox后,car-t细胞几乎对csf1r阳性细胞不产生杀伤作用,不再分泌细胞因子il-2和ifn-γ。采用本发明的car-t细胞能调控性杀伤靶细胞,能够控制细胞因子分泌水平,有效避免了car-t细胞发挥治疗作用可能触发的“细胞因子风暴”副作用。
[0111]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。