1.本发明属于医药技术领域,涉及4-甲氧基萘取代的苯胺类化合物及其制备方法和应用。
背景技术:
2.胰岛素抵抗,高血脂症,脂肪肝都是导致血脑血管疾病的原因,是威胁人类健康与生命的严重疾病,寻找和发现治疗与预防心脑血管疾病相关的新药是当前面临的重大课题。
3.组成型雄甾烷受体(car,nr1i3)和孕烷x受体(pxr,nr1i2)是核受体超家族的两个重要成员,在异源化学刺激中起关键作用。car诱导各种药物代谢酶(例如cyp2b6,cyp3a4,cyp2c9和ugt1a1)和转运蛋白的表达。car通常被隔离在未经处理的肝细胞的胞质区中,并在暴露于激活剂或配体后易位至细胞核。核易位后,car与其靶基因启动子区域的响应元件结合,与类维生素ax受体α(rxrα,nr2b1)形成异二聚体复合物。相反,外源表达的car在细胞核中积累而没有任何刺激。因为car具有组成型反式激活的潜力,所以在没有激动剂的情况下,核car会诱导靶基因转录。据报道,car在能量稳态中起重要作用,例如影响甲状腺激素代谢,葡萄糖生成和脂肪生成进而对心脑血管疾病产生治疗作用。多数car激动剂以睾丸酮为原料合成,存在副作用大,合成复杂,易使其他官能团受影响的缺点。
4.4-甲氧基萘取代的苯胺类化合物在作为car激动剂方面的研究尚未见报道。
技术实现要素:
5.本发明的目的在于设计、合成具有良好car激动剂作用的4-甲氧基萘取代的苯胺类化合物,所制备的化合物在体外具有良好的car激动剂作用。
6.本发明涉及定义如下的通式m的化合物或其药学上可接受的盐:
[0007][0008]
其中,
[0009]
r1为氢、c1-c6烷氧基、卤素、c1-c6烷基、羟基、硝基、氨基;
[0010]
r2为氢、c1-c6烷基、卤素、c1-c6烷氧基。
[0011]
本发明优选定义如下的通式m的化合物或其药学上可接受的盐,其中,
[0012]
r1为氢、c1-c4烷氧基、卤素、c1-c4烷基、羟基、硝基、氨基;
[0013]
r2为氢、c1-c4烷基、卤素、c1-c4烷氧基。
[0014]
本发明优选定义如下的通式m的化合物或其药学上可接受的盐,其中,
[0015]
r1为氢、c1-c4烷氧基、卤素;
[0016]
r2为氢、c1-c4烷基、卤素;
[0017]
本发明优选定义如下的通式m的化合物或其药学上可接受的盐:其中,
[0018]
r1为氢时,r2为氢、卤素、c1-c4烷基;
[0019]
r1为c1-c4烷氧基时,r2为氢、c1-c4烷基、卤素;
[0020]
r1为卤素时,r2为氢、c1-c4烷基、卤素;
[0021]
本发明优选定义如下的通式m的化合物或其药学上可接受的盐:其中,
[0022]
r1为氢时,r2为h,me,cl,f;
[0023]
r1为甲氧基时,r2为h,me,cl,f;
[0024]
r1为氯、氟时,r2为h,me,cl,f;
[0025]
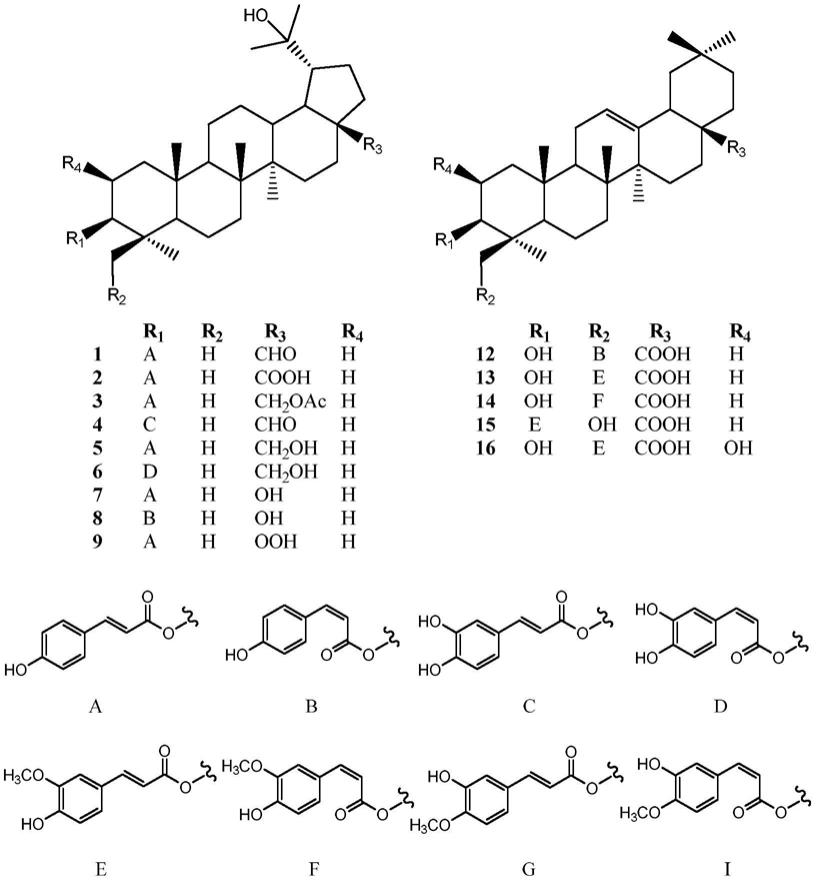
本发明优选的部分化合物结构如下:
[0026]
化合物1
[0027][0028]
化合物2
[0029][0030]
化合物3
[0031][0032]
化合物4
[0033][0034]
化合物5
[0035][0036]
化合物6
[0037][0038]
化合物7
[0039][0040]
化合物8
[0041][0042]
化合物9
[0043][0044]
化合物10
[0045]
[0046]
化合物11
[0047][0048]
化合物12
[0049][0050]
化合物13
[0051][0052]
化合物14
[0053][0054]
化合物15
[0055][0056]
化合物16
[0057][0058]
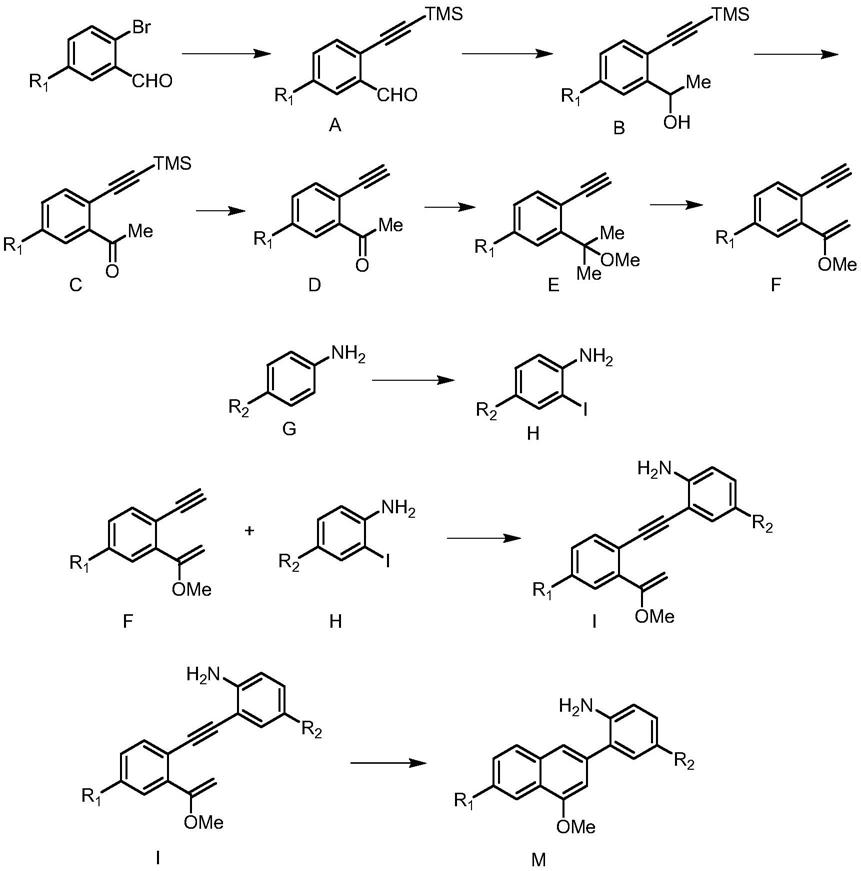
本发明的4-甲氧基萘取代的苯胺类化合物合成路线如下:
[0059]
化合物a的制备
[0060]
先向装有2-溴代-5-取代苯甲醛,双三苯基膦二氯化钯,碘化亚铜的茄型瓶中加入适量无氧且干燥的四氢呋喃,在氮气保护的条件下,加入三甲基乙炔基硅和干燥三乙胺,并继续搅拌直至末端炔烃完全被消耗。待反应结束后,将反应液浓缩并柱层析,得化合物a。
[0061]
化合物b的制备
[0062]
将化合物a加入到干燥的茄型瓶中,加入干燥的乙醚,氮气保护,在-78℃条件下滴加甲基溴化镁,加毕,将反应液升至室温,继续搅拌2-4小时。待反应结束后,加入饱和氯化铵溶液淬灭,乙酸乙酯萃取,水洗,硫酸钠干燥,浓缩得化合物b。
[0063]
化合物c的制备
[0064]
将pcc加入到干燥的茄型瓶中,加入干燥的二氯甲烷,氮气保护,0℃条件下加入化合物b的二氯甲烷溶液,加毕,将反应液升至室温,搅拌12小时以上或过夜。反应结束后,将反应液过砂氏漏斗,得化合物c。
[0065]
化合物d的制备
[0066]
向茄型瓶中加入化合物c,碳酸钾和甲醇,室温搅拌2-4小时。反应结束后,加入饱和氯化铵溶液淬灭,二氯甲烷萃取,水洗,硫酸钠干燥,浓缩得化合物d。
[0067]
化合物e的制备
[0068]
向茄型瓶中加入化合物d,原甲酸三甲酯,对甲苯磺酸,甲醇,室温搅拌3-5小时。待反应结束后,加入三乙胺淬灭,乙酸乙酯萃取,饱和碳酸氢钠溶液洗,水洗,硫酸钠干燥,浓缩得化合物e。
[0069]
化合物f的制备
[0070]
向茄型瓶中加入化合物e,三甲基氯硅烷,苯甲酸,吡啶,70-90℃搅拌10小时以上或过夜。反应结束后,0℃下向反应液中加入15%naoh,乙酸乙酯萃取,硫酸铜溶液洗,水洗,硫酸钠干燥,浓缩后柱层析,得化合物f。
[0071]
化合物h的制备
[0072]
将化合物g溶于水-甲醇溶液(100:3)中,加入碳酸氢钠,碘单质,室温搅拌20-40分钟。反应结束后,加入水稀释,并用盐酸(2m)将ph调中性,乙醚萃取,硫代硫酸钠洗,水洗,硫酸钠干燥,浓缩后柱层析得化合物h。
[0073]
化合物i的制备
[0074]
先向装有化合物h,双三苯基膦二氯化钯,碘化亚铜的茄型瓶中加入适量无氧且干燥的四氢呋喃,在氮气保护的条件下,加入化合物f和干燥三乙胺,并继续搅拌直至末端炔烃完全被消耗。待反应结束后,将反应液浓缩并柱层析,得化合物i。
[0075]
化合物m的制备
[0076]
向干燥的茄型瓶内加入ipraucl和agsbf6,干燥二氯甲烷溶解,常温搅拌20-40分钟,加入化合物i的二氯甲烷溶液,继续搅拌2-4小时。待反应结束后,浓缩反应液,柱层析,得化合物m。
[0077]
本发明还提供了一种药物组合物,包含所述的4-甲氧基萘取代的苯胺类化合物或其盐和药学上可接受的载体或赋形剂。
[0078]
本发明的4-甲氧基萘取代的苯胺类化合物或其盐具有良好的car激动剂的作用。
附图说明
[0079]
图1为各化合物用citco处理后对car受体的激动率
具体实施方式
[0080]
无水四氢呋喃和无水乙醚是通过在氮气存在的条件下加入钠条回流蒸出制得的。无水二氯甲烷则是通过在氮气存在的条件下加入氢化钙回流蒸出制得的。所涉及到的化学反应都是用f-254薄层硅胶板进行的tlc分析。反应液的提取与分离则都是用200-300目的硅胶为填充剂进行柱层析实现的。1h nmr和
13
c nmr是氘代二甲基亚砜为溶剂利用bruker avance-iii 600进行测试得到的。
[0081]
实施例1:2-(4-甲氧基萘-2-基)苯胺(化合物1)的制备
[0082]
先向装有邻溴苯甲醛(5g,0.0272mol),双三苯基膦二氯化钯(0.95g,0.0014mol),碘化亚铜(0.50g,0.0026mol)的茄型瓶中加入适量无氧且干燥的四氢呋喃,在氮气保护的条件下,加入三甲基乙炔基硅(6.67g,0.0679mol)和干燥三乙胺(19.1ml,0.136mol),并继续搅拌直至末端炔烃完全被消耗。待反应结束后,将反应液浓缩并柱层析,得2-((三甲基甲硅烷基)乙炔基)苯甲醛,收率99%。
[0083]
将2-((三甲基甲硅烷基)乙炔基)苯甲醛(5.49g,0.027mol)加入到干燥的茄型瓶中,加入干燥的乙醚,氮气保护,在-78℃条件下滴加甲基溴化镁(9.66g,0.081mol),加毕,将反应液升至室温,继续搅拌3小时。待反应结束后,加入饱和氯化铵溶液淬灭,乙酸乙酯萃取,水洗,硫酸钠干燥,浓缩得1-(2-((三甲基甲硅烷基)乙炔基)苯基)乙-1-醇,收率99%。
[0084]
将pcc(9.52g,0.041mol)加入到干燥的茄型瓶中,加入干燥的二氯甲烷,氮气保护,0℃条件下加入1-(2-((三甲基甲硅烷基)乙炔基)苯基)乙-1-醇(6.19g,0.027mol)的二氯甲烷溶液,加毕,将反应液升至室温,搅拌过夜。反应结束后,将反应液过砂氏漏斗,得1-(2-((三甲基甲硅烷基)乙炔基)苯基)乙-1-酮,收率99%。
[0085]
向茄型瓶中加入1-(2-((三甲基甲硅烷基)乙炔基)苯基)乙-1-酮(5.83g,0.027mol),碳酸钾(0.37g,0.0027mol)和甲醇,室温搅拌3小时。反应结束后,加入饱和氯化铵溶液淬灭,二氯甲烷萃取,水洗,硫酸钠干燥,浓缩得1-(2-乙炔基苯基)乙-1-酮,收率99%。
[0086]
向茄型瓶中加入1-(2-乙炔基苯基)乙-1-酮(3.89g,0.027mol),原甲酸三甲酯(7.16g,0.0675mol),对甲苯磺酸(0.94g,0.0054mol),甲醇,室温搅拌4小时。待反应结束后,加入三乙胺淬灭,乙酸乙酯萃取,饱和碳酸氢钠溶液洗,水洗,硫酸钠干燥,浓缩得1-乙炔基-2-(2-甲氧基丙-2-基)苯,收率95%
[0087]
向茄型瓶中加入1-乙炔基-2-(2-甲氧基丙-2-基)苯(4.47g,0.026mol),三甲基氯硅烷(7.18ml,0.065mol),苯甲酸(0.57g,0.00468mol),吡啶,80℃搅拌过夜。反应结束后,0℃下向反应液中加入15%naoh,乙酸乙酯萃取,硫酸铜溶液洗,水洗,硫酸钠干燥,浓缩后柱层析,得1-乙炔基-2-(1-甲氧基乙烯基)苯,收率86%。
[0088]
将苯胺(5g,0.054mol)溶于水-甲醇溶液(100:3)中,加入碳酸氢钠,碘单质(10.9g,0.0432mol),室温搅拌30分钟。反应结束后,加入水稀释,并用盐酸(2m)将ph调至7,乙醚萃取,硫代硫酸钠洗,水洗,硫酸钠干燥,浓缩后柱层析得邻碘苯胺,收率90%。
[0089]
先向装有邻碘苯胺(0.46g,1.58mmol),双三苯基膦二氯化钯(0.022g,0.032mmol),碘化亚铜(0.011g,0.063mmol)的茄型瓶中加入适量无氧且干燥的四氢呋喃,在氮气保护的条件下,加入1-乙炔基-2-(1-甲氧基乙烯基)苯(0.1g,0.63mmol)和干燥三乙胺(0.44ml,3.15mmol),并继续搅拌直至末端炔烃完全被消耗。待反应结束后,将反应液浓缩并柱层析,得2-((2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺,收率92%。
[0090]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入2-((2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(25mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物1,收率92%。1h nmr(600mhz,dmso-d6)δ8.14(d,j=8.2hz,1h),7.89(d,j=8.0hz,1h),7.55
–
7.51(m,1h),7.51
–
7.46(m,2h),7.13(dd,j=7.5,1.5hz,1h),7.10
–
7.06(m,1h),6.99(d,j=1.1hz,1h),6.79(dd,j=8.0,0.9hz,1h),6.67(td,j=7.4,1.1hz,1h),4.93(d,j=16.7hz,2h),4.00(s,3h);
13
c nmr(150mhz,dmso-d6)δ155.30,145.70,138.18,134.69,130.66,128.74,128.18,127.13,126.32,125.64,124.29,121.76,119.83,117.05,115.69,106.22,56.03。
[0091]
实施例2:2-(4-甲氧基萘-2-基)-4-甲基苯胺(化合物2)的制备
[0092]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((2-(1-甲氧基乙烯基)苯基)乙炔基)-4-甲基苯胺(26mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物2,收率93%。1h nmr(600mhz,dmso-d6)δ8.14(d,j=8.3hz,1h),7.89(d,j=8.0hz,1h),7.55
–
7.51(m,1h),7.50
–
7.45(m,2h),7.00
–
6.94(m,2h),6.90(dd,j=8.1,1.7hz,1h),6.71(d,j=8.1hz,1h),4.70(s,2h),4.00(s,3h),2.21(s,3h);
13
c nmr(150mhz,dmso-d6)δ155.24,143.19,138.32,134.68,131.03,129.27,128.15,127.11,126.40,125.59,125.46,124.24,121.76,119.80,115.95,106.23,56.04,20.59。
[0093]
实施例3:4-氯-2-(4-甲氧基萘-2-基)苯胺(化合物3)的制备
[0094]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氯-2-((2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(28mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物3,收率87%。1h nmr(600mhz,dmso-d6)δ7.99(dd,j=9.0,5.8hz,1h),7.74(td,j=10.7,2.7hz,1h),7.54(s,1h),7.48
–
7.41(m,1h),7.04(s,1h),6.95(s,1h),6.92
–
6.88(m,1h),6.71(d,j=8.1hz,1h),4.71(s,2h),4.00(s,3h),2.21(s,3h);
13
c nmr(150mhz,dmso-d6)δ155.45,144.92,136.74,134.63,129.78,128.31,127.71,127.22,125.90,124.51,121.78,120.05,117.08,105.88,56.09。
[0095]
实施例4:4-氟-2-(4-甲氧基萘-2-基)苯胺(化合物4)的制备
[0096]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氟-2-((2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(27mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物4,收率90%。1h nmr(600mhz,dmso-d6)δ8.15(d,j=8.3hz,1h),7.90(d,j=7.9hz,1h),7.57
–
7.48(m,3h),7.02
–
6.97(m,2h),6.94(td,j=8.6,3.0hz,1h),6.79(dd,j=8.8,5.2hz,1h),4.83(s,2h),4.01(s,3h);
13
c nmr(150mhz,dmso-d6)δ155.41,155.06(d,j=229.5hz),142.33,137.01,134.58,128.29,127.23,127.04(d,j=7.1hz),125.90,124.45,121.77,119.99,116.63,116.53(d,j=15.8hz),115.17(d,j=21.8hz),105.96,56.09。
[0097]
实施例5:2-(4,6-二甲氧基萘-2-基)苯胺(化合物5)的制备
[0098]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-甲氧基-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(28mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物5,收率94%。1h nmr(600mhz,dmso-d6)δ7.82(d,j=8.9hz,1h),7.47
–
7.43(m,2h),7.19(dt,j=10.6,5.3hz,1h),7.12(dd,j=
7.5,1.4hz,1h),7.08
–
7.04(m,1h),6.98(d,j=1.0hz,1h),6.82
–
6.76(m,1h),6.66(td,j=7.4,1.0hz,1h),4.88(s,2h),4.00(s,3h),3.88(s,3h);
13
c nmr(150mhz,dmso-d6)δ157.47,154.43,145.68,135.56,130.66,129.96,128.53,126.47,125.19,119.73,119.41,117.07,115.64,106.68,100.31,55.96,55.62。
[0099]
实施例6:2-(4,6-二甲氧基萘-2-基)-4-甲基苯胺(化合物6)的制备
[0100]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-甲氧基-2-(1-甲氧基乙烯基)苯基)乙炔基)-4-甲基苯胺(29mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物6,收率89%。1h nmr(600mhz,dmso-d6)δ7.81(d,j=8.9hz,1h),7.46
–
7.41(m,2h),7.18(dd,j=8.9,2.6hz,1h),6.95(dd,j=17.5,1.4hz,2h),6.88(dd,j=8.1,1.7hz,1h),6.69(d,j=8.1hz,1h),4.66(s,2h),3.99(s,3h),3.88(s,3h),2.20(s,3h);
13
c nmr(150mhz,dmso-d6)δ157.43,154.38,143.15,135.70,131.05,129.93,129.05,126.55,125.46,125.14,119.70,119.39,115.89,106.68,100.31,55.97,55.61,20.59。
[0101]
实施例7:4-氯-2-(4,6-二甲氧基萘-2-基)苯胺(化合物7)的制备
[0102]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氯-2-((4-甲氧基-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(31mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物7。收率95%。1h nmr(600mhz,dmso-d6)δ7.84(d,j=8.9hz,1h),7.48
–
7.43(m,2h),7.20(dd,j=8.9,2.6hz,1h),7.13
–
7.07(m,2h),6.96(d,j=1.0hz,1h),6.79(d,j=8.6hz,1h),5.07(s,2h),4.00(d,j=5.4hz,3h),3.89(s,3h);
13
c nmr(100mhz,dmso-d6)δ157.67,154.57,144.88,134.12,130.03,129.92,129.77,128.10,127.89,125.47,120.09,119.90,119.49,117.01,106.33,100.37,56.04,55.65。
[0103]
实施例8:2-(4,6-二甲氧基萘-2-基)-4-氟苯胺(化合物8)的制备
[0104]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氟-2-((4-甲氧基-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(30mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物8。收率94%。1h nmr(600mhz,dmso-d6)δ7.83(d,j=8.9hz,1h),7.48(s,1h),7.45(d,j=2.6hz,1h),7.20(dd,j=8.9,2.6hz,1h),7.00
–
6.95(m,2h),6.95
–
6.90(m,1h),6.78(dd,j=8.8,5.2hz,1h),4.82(s,2h),4.00(d,j=6.3hz,3h),3.88(s,3h);
13
c nmr(150mhz,dmso-d6)δ157.65,155.11(d,j=229.5hz),154.52,142.23,134.40,130.03,129.87,125.40,119.88,119.51,116.61,116.54,116.47,114.92(d,j=21.7hz),106.40,100.31,56.02,55.64(s)。
[0105]
实施例9:2-(6-氯-4-甲氧基萘-2-基)苯胺(化合物9)的制备
[0106]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-氯-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(28mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物9。收率94%。1h nmr(600mhz,dmso-d6)δ
8.10(d,j=2.1hz,1h),7.96(d,j=8.8hz,1h),7.55(dd,j=8.7,1.9hz,2h),7.15
–
7.05(m,3h),6.82
–
6.78(m,1h),6.67(td,j=7.4,1.0hz,1h),4.95(s,2h),4.01(s,3h);
13
c nmr(150mhz,dmso-d6)δ154.47,145.74,138.91,133.07,130.67,130.53,130.33,128.92,127.56,125.88,124.86,120.66,119.73,117.06,115.77,107.49,56.23。
[0107]
实施例10:2-(6-氯-4-甲氧基萘-2-基)-4-甲基苯胺(化合物10)的制备
[0108]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-氯-2-(1-甲氧基乙烯基)苯基)乙炔基)-4-甲基苯胺(30mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物10。收率92%。1h nmr(600mhz,dmso-d6)δ8.10(d,j=2.1hz,1h),7.94(d,j=8.8hz,1h),7.53(dd,j=7.9,3.0hz,2h),7.06(d,j=0.9hz,1h),6.96(d,j=1.5hz,1h),6.91(dd,j=8.1,1.7hz,1h),6.72(d,j=8.1hz,1h),4.73(s,2h),4.00(s,3h),2.21(s,3h);
13
c nmr(150mhz,dmso-d6)δ154.42,143.23,139.06,133.06,131.02,130.47,130.28,129.47,127.53,125.99,125.51,124.82,120.67,119.70,116.05,107.48,56.22,20.56。
[0109]
实施例11:4-氯-2-(6-氯-4-甲氧基萘-2-基)苯胺(化合物11)的制备
[0110]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氯-2-((4-氯-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(32mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物11。收率93%。1h nmr(600mhz,dmso-d6)δ8.10(d,j=2.2hz,1h),7.97(d,j=8.8hz,1h),7.58
–
7.55(m,2h),7.16
–
7.09(m,2h),7.05(d,j=1.2hz,1h),6.80(d,j=8.6hz,1h),5.14(s,2h),4.02(d,j=9.7hz,3h);
13
c nmr(150mhz,dmso-d6)δ154.61,144.93,137.43,133.00,130.61,129.79,128.50,127.64,127.27,125.09,120.70,120.14,119.92,117.17,107.13,56.28。
[0111]
实施例12:2-(6-氯-4-甲氧基萘-2-基)-4-氟苯胺(化合物12)的制备
[0112]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-氯-2-(1-甲氧基乙烯基)苯基)乙炔基)-4-氟苯胺(30mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物12。收率97%。1h nmr(600mhz,dmso-d6)δ8.10(d,j=2.1hz,1h),7.95(t,j=9.5hz,1h),7.60
–
7.53(m,2h),7.08(d,j=1.0hz,1h),7.01
–
6.92(m,2h),6.79(dd,j=8.8,5.2hz,1h),4.86(s,2h),4.02(s,3h);
13
c nmr(150mhz,dmso-d6)δ155.06(d,j=229.5hz),154.57,142.37(d,j=1.1hz),137.73,132.96,130.62,130.60,127.66,126.58(d,j=7.0hz),125.03,120.68,119.91,116.72(d,j=7.5hz),116.56(d,j=22.1hz),115.37(d,j=21.8hz),107.24,56.29。
[0113]
实施例13:2-(6-氟-4-甲氧基萘-2-基)苯胺(化合物13)的制备
[0114]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-氟-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(27mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物13。收率94%。1h nmr(600mhz,dmso-d6)δ8.10(d,j=2.1hz,1h),7.95(t,j=9.5hz,1h),7.60
–
7.53(m,2h),7.08(d,j=1.0hz,1h),
7.01
–
6.92(m,2h),6.79(dd,j=8.8,5.2hz,1h),4.86(s,2h),4.02(s,3h);
13
c nmr(150mhz,dmso-d6)δ160.28(d,j=242.9hz),154.81(d,j=5.3hz),145.74,137.63(d,j=2.3hz),131.84,131.21(d,j=8.8hz),130.68,128.80,126.04,124.85(d,j=8.7hz),119.84,117.09(d,j=25.5hz),117.05,115.72,107.27,105.44(d,j=22.2hz),56.17。
[0115]
实施例14:2-(6-氟-4-甲氧基萘-2-基)-4-甲基苯胺(化合物14)的制备
[0116]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得2-((4-氟-2-(1-甲氧基乙烯基)苯基)乙炔基)-4-甲基苯胺(28mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物14。收率95%。1h nmr(600mhz,dmso-d6)δ7.99(dd,j=9.0,5.8hz,1h),7.74(td,j=10.7,2.7hz,1h),7.54(s,1h),7.48
–
7.41(m,1h),7.04(s,1h),6.95(s,1h),6.92
–
6.88(m,1h),6.71(d,j=8.1hz,1h),4.71(s,2h),4.00(s,3h),2.21(s,3h).
13
c nmr(150mhz,dmso-d6)δ160.25(d,j=242.9hz),154.74(d,j=5.2hz),143.22,137.78(d,j=2.5hz),131.82,131.18(d,j=8.8hz),131.05,129.34,126.11,125.46,124.79(d,j=8.7hz),119.80,117.07(d,j=25.0hz),115.98,107.26,105.43(d,j=22.2hz),56.17,20.57。
[0117]
实施例15:4-氯-2-(6-氟-4-甲氧基萘-2-基)苯胺(化合物15)的制备
[0118]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氯-2-((4-氟-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(30mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物15。收率96%。1h nmr(600mhz,dmso-d6)δ8.01(dd,j=9.0,5.8hz,1h),7.76(dd,j=10.6,2.5hz,1h),7.58(d,j=11.6hz,1h),7.47(td,j=8.8,2.7hz,1h),7.14
–
7.08(m,1h),7.03(s,1h),6.80(d,j=8.6hz,1h),5.12(s,1h),4.01(s,2h);
13
c nmr(150mhz,dmso-d6)δ160.42(d,j=243.3hz),154.92(d,j=5.1hz),144.96,136.18,131.77,131.37(d,j=8.9hz),129.80,128.3,127.38,125.10(d,j=8.8hz),120.03,117.20(d,j=25.0hz),117.09,106.93,105.47(d,j=22.2hz),56.24。
[0119]
实施例16:4-氟-2-(6-氟-4-甲氧基萘-2-基)苯胺(化合物16)的制备
[0120]
向干燥的茄型瓶内加入ipraucl(3.0mg,0.005mmol)和agsbf6(1.72mg,0.005mmol),干燥二氯甲烷溶解,常温搅拌30分钟,加入以实施例1相同的方法制得4-氟-2-((4-氟-2-(1-甲氧基乙烯基)苯基)乙炔基)苯胺(29mg,0.1mmol)的二氯甲烷溶液,继续搅拌2小时。待反应结束后,浓缩反应液,柱层析,得化合物16。收率96%。1h nmr(600mhz,dmso-d6)δ8.01(dd,j=9.0,5.7hz,1h),7.76(dd,j=10.6,2.6hz,1h),7.60(s,1h),7.47(td,j=8.8,2.7hz,1h),7.06(s,1h),7.01
–
6.92(m,2h),6.79(dd,j=8.8,5.2hz,1h),4.84(s,2h),4.01(s,3h);
13
c nmr(150mhz,dmso-d6)δ160.41(d,j=243.2hz),155.04(d,j=231.0hz),154.89(d,j=5.2hz),142.37,136.47,131.73,131.35(d,j=8.9hz),126.73(d,j=7.0hz),125.05(d,j=8.8hz),120.01,117.21(d,j=25.0hz),116.65(d,j=2.1hz),116.56(d,j=16.8hz),115.24(d,j=21.8hz),107.00,105.47(d,j=22.2hz),56.23。
[0121]
实施例17
[0122]
本发明化合物体外作为car激动剂的活性测试
[0123]
细胞培养先前使用t-rex系统(invitrogen/thermo fisher scientific,waltham,ma)建立了源自人类肝癌hepg2细胞的heptr/hcar细胞系,其中可以通过tet处理诱导hcar的表达。通过用pcmv3tag6-hpxr转染建立了稳定表达hpxr的人肝癌hepg2细胞系(hep/hpxr)。将heptr/hcar hepg2和hep/hpxr细胞在含有10%胎牛血清,青霉素和链霉素的dulbecco改良版eagle培养基(日本wako,日本大阪)中,在5%co2的潮湿环境中于37℃培养。将分化的heparg细胞(kac,日本京都)保存在威廉姆斯培养基e(life technologies/thermo fisher scientific,纽约格兰德岛)中,并补充10%胎牛血清,5mg/ml胰岛素和50mm氢化可的松(wako)(日本大阪)),根据制造商的说明,在37℃,5%的co2和95%的空气中进行。
[0124]
质粒gal/hcar lbd,gal/hcar lbd sv2和gal/hcar lbd 3a.a的表达质粒。(kanno and inouye,2010 35 515-525)(2013 38 309-315)。先前已构建了gal响应元件(galre)/tata盒驱动的高斯荧光素酶报道载体(gal-gaussia luc。)(2019 102977 88)。先前已经描述了cyp3a4 xrem驱动的萤光素酶报告质粒(xremluc。)的制备(2019 95 120-126)。
[0125]
用gal/hcar lbd 3a.a的表达载体转染高斯荧光素酶试验hepg2细胞。使用pei max reagent(polysciences inc.,warrington,pa)和galre驱动的高斯荧光素酶报告载体。过夜孵育后,将细胞用此专利中化合物(10um)或citcio(1um)处理24小时,并使用biolux高斯荧光素酶测定试剂盒(biorad,hercules,ca)测量高斯荧光素酶活性。
[0126]
用gal/hcar lbd,gal/hcar lbd sv2,gal/hcar lbd 3a.a.,pg5luc和pgl4.74(hrluc/tk;promega,madison,wi);我们使用了pei max reagent(polysciences inc.)。同样,用cyp3a4 xrem驱动的荧光素酶报告质粒和pgl4.74转染hep/hpxr细胞。过夜孵育后,用化合物处理细胞,并使用双重萤光素酶报告基因测定系统(promega)测量萤光素酶活性。萤火虫荧光素酶的活性被标准化为海肾荧光素酶的活性。
[0127]
定量逆转录聚合酶链反应(rt-pcr)使用isogen ii(日本基因,东京,日本)分离来自heptr/hcar,hep/hpxr和heparg细胞的总rna,并使用revertraace qpcr rt试剂盒(toyobo)合成cdna。日本大阪)。根据制造商的协议,使用geneace sybr qpcr mixαlow rox(日本基因,东京,日本)在7500fast系统(applied biosystems,foster city,ca)上进行定量pcr(qpcr)。所使用的特异性pcr引物如下:cyp2b6(5'-aag cgg att tgt ctt ggt gaa-3'和5'-tggagg atg gtg gtg aag aag3'),cyp3a4(5'-cca agc tat gct ctt cac cg-3'和5'-tcaggc tcc act tac ggt gc-3')和b-肌动蛋白(5'-tcc tcctga gcg caa gta ctc-3'和5'ctg ctt gct gat cca cat ctg-3')。统计分析进行统计比较时,先采用方差单向分析,然后使用dunnett的多重比较检验作为事后检验,并且差异被认为具有统计学意义,*p《0.05。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。