1.本发明涉及溶液相肽核酸(pna)寡聚体的制备方法。
背景技术:
2.众所周知,核酸为担当生物的遗传信息的脱氧核糖核酸(dna)及核糖核酸(rna)。
3.相反,肽核酸为将核酸的糖磷酸骨架转换为n-(2-氨基乙基)甘氨酸骨架的核酸。
4.脱氧核糖核酸/核糖核酸的糖磷酸骨架在中性条件下因具有负电荷而使互补链(complementary chains)之间存在静电斥力,但肽核酸的骨架结构(backbone structure)原本不具有电荷,因此没有静电斥力。
5.即,肽核酸主链实质上完全不带电荷,这是肽核酸非常重要的特征,由于上述特征,可以将其用在许多无法使用寡核苷酸或寡核苷酸衍生物的用途中。
6.进而,肽核酸以比寡核苷酸更高的亲和性与脱氧核糖核酸或核糖核酸结合,与天然核糖核酸相比,具有在血清中非常稳定的优点。
7.然而,虽然对肽核酸应用的关注和研究很高且多种多样,但有关肽核酸寡聚体的合成方法的研究却很迟缓。
8.通常,肽核酸寡聚体的合成方法利用固相(solid-phase)肽合成法,若以肽核酸的骨架结构对肽核酸单体单元(unit)进行分类,则可以分为fmoc型肽核酸单体单元和boc型肽核酸单体单元两种。
9.虽然已经确立了fmoc型肽核酸单体单元的合成方法,但不易大量生产,不仅如此,还存在收率及纯度的问题。
10.例如,虽然wo2005-009998a1公开了易于以高收率合成肽核酸的单体,但因多个工序而在大量生产中存在不经济的问题。
11.因此,需要能够通过简单的工序来有效地以高纯度及收率来制备目标肽核酸的溶液相肽核酸寡聚体的制备方法。
技术实现要素:
技术问题
12.本发明提供通过简单的工序以惊人地提高的纯度及收率制备目标肽核酸寡聚体的方法。技术方案
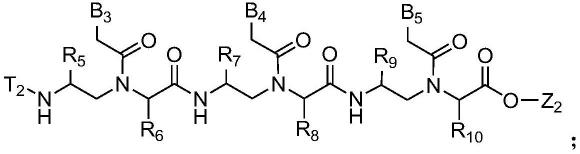
13.本发明提供能够通过简单的工序、容易的溶液相以极度提高的纯度及收率制备肽核酸寡聚体的方法,本发明的肽核酸寡聚体的制备方法的特征在于,包括在溶液相中使肽核酸嵌段与肽核酸嵌段反应来制备肽核酸寡聚体的步骤,上述肽核酸嵌段由不同或相同的两个以上的肽核酸单体结合而成。
14.优选地,本发明的肽核酸寡聚体的制备方法包括在存在偶联试剂及溶剂的情况下,将由下述化学式1表示的第一肽核酸二聚体、由下述化学式2表示的第一肽核酸三聚体
或由下述化学式3表示的第一肽核酸四聚体与由下述化学式4表示的第二肽核酸二聚体、由下述化学式5表示的第二肽核酸三聚体或由下述化学式6表示的第二肽核酸四聚体混合后加入胺类化合物来制备由下述化学式7至化学式9表示的肽核酸寡聚体的步骤。
15.化学式1
16.化学式2
17.化学式3
18.化学式4
19.化学式5
20.化学式6
21.化学式7
22.化学式8
23.化学式9
24.在上述化学式1至化学式9中,a1至a9及a
11
至a
14
各自独立地为包含相互不同或相同的核酸碱基的肽核酸单体,p1至p18各自独立地为氢,或者相互不同或相同的保护基,p1与p2、p3与p4、p5与p6、p
11
与p
12
、p
13
与p
14
、p
15
与p
16
及p
17
与p
18
各自同时为氢的情况除外,a及b各自独立地为1的整数,c及d各自独立地为0或1的整数。
25.优选地,本发明一实施例的肽核酸寡聚体的制备方法包括:步骤a),在存在偶联试剂及溶剂的情况下向由下述化学式1-1表示的第一肽核酸二聚体、由下述化学式2-1表示的第一肽核酸三聚体或由下述化学式3-1表示的第一肽核酸四聚体加入由下述化学式4-1表示的第二肽核酸二聚体、由下述化学式5-1表示的第二肽核酸三聚体或由下述化学式6-1表示的第二肽核酸四聚体来获得混合液;以及步骤b),向上述步骤的混合液加入胺类化合物来制备上述化学式7至化学式8的化合物。
26.化学式1-1
27.化学式2-1
28.化学式3-1
29.化学式4-1
30.化学式5-1
31.化学式6-1
32.在上述化学式1-1、化学式2-1、化学式3-1、化学式4-1、化学式5-1及化学式6-1中,a1至a9及a
11
至a
14
各自独立地为包含相互不同或相同的核酸碱基的肽核酸单体,p2、p4、p6、p7、p9及p
11
各自独立地为氢或相互不同或相同的保护基,x1至x3各自独立地为酸性盐,a及b各自独立地为1的整数,c及d各自独立地为0或1的整数。
33.优选地,本发明一实施例的胺类化合物可以为n,n-二异丙基乙胺,但不限定于此。
34.优选地,本发明一实施例的肽核酸寡聚体的制备方法还可以包括再次利用上述步骤b)中获得的产物作为起始物质来重复执行步骤a)及步骤b)的步骤。
35.更优选地,混合液内的起始物质与溶剂的体积比可以为1∶10以上。
36.本发明一实施例的核酸碱基可以为腺嘌呤、胞嘧啶、5-甲基胞嘧啶、鸟嘌呤、胸腺嘧啶、尿嘧啶、嘌呤、2,6-二氨基嘌呤、n4n
4-桥亚乙基胞嘧啶、n6n
6-桥亚乙基-2,6-二氨基嘌呤、5-(c3-c6)-炔基尿嘧啶、5-(c3-c6)-炔基-胞嘧啶、5-(1-炔丙基氨基)尿嘧啶、5-(1-炔丙基氨基)胞嘧啶、吩恶嗪、9-氨基乙氧基吩恶嗪、5-氟尿嘧啶、假异胞嘧啶、5-(羟甲基)尿
二异丙基乙胺(diea)的混合溶剂。
46.本发明的一实施例还可以包括向化学式7至化学式9的化合物加入水来分离纯化的步骤。发明的效果
47.本发明的肽核酸寡聚体的制备方法通过制备肽核酸嵌段后使肽核酸嵌段与肽核酸嵌段反应来制备肽核酸寡聚体,划时代地减少肽核酸的制备步骤,可以在非常经济的同时以高纯度制备肽核酸寡聚体。
48.具体地,本发明的肽核酸寡聚体的制备方法使用肽核酸二聚体、肽核酸三聚体或肽核酸四聚体来制备,与利用肽核酸单体制备的现有的方法相比,能够以更为简单的工序制备肽核酸寡聚体,不仅如此,还可以更为正确地制备目标肽核酸寡聚体。
49.因此,与利用肽核酸单体制备的现有的方法相比,本发明的肽核酸寡聚体的制备方法以更短的工序步骤制备,与副产物的分离非常容易,因此肽核酸寡聚体的收率及纯度非常高。
50.进而,本发明的肽核酸寡聚体的制备方法的所有工序都可以在溶液相中制备,能够大量生产,因此非常有利于商业化。
51.并且,本发明的肽核酸寡聚体的制备方法使用肽核酸嵌段,具体地,使用肽核酸二聚体、肽核酸三聚体或肽核酸四聚体来制备,与现有的方法相比,使用非常少量的肽核酸二聚体、肽核酸三聚体或肽核酸四聚体,因此非常经济,能够以高收率轻松地以大量生产的方式制备高纯度的肽核酸寡聚体。
附图说明
52.图1为本发明的实施例20中制备的肽核酸寡聚体的高效液相色谱法(hplc)测量结果。
53.图2为本发明的比较例1中制备的肽核酸寡聚体的高效液相色谱法测量结果。
具体实施方式
54.以下,详细说明本发明的肽核酸寡聚体的制备方法及根据该方法制备的肽核酸寡聚体,在此情况下,若无其他定义,则所使用的技术术语及科学术语具有本发明所属技术领域的普通技术人员通常理解的含义,在下述说明中,将省略可能不必要地混淆本发明的要旨的公知的功能及结构。
55.本说明书所记载的氨基酸以广泛的含义来使用,不仅包括天然氨基酸,例如丝氨酸(ser)、天冬酰胺(asn)、缬氨酸(val)、亮氨酸(leu)、异亮氨酸(ile)、丙氨酸(ala)、酪氨酸(tyr)、甘氨酸(gly)、赖氨酸(lys)、精氨酸(arg)、组氨酸(his)、天冬氨酸(asp)、谷氨酸(glu)、谷氨酰胺(gln)、苏氨酸(thr)、半胱氨酸(cys)、蛋氨酸(met)、苯丙氨酸(phe)、色氨酸(trp)、脯氨酸(pro),还包括氨基酸变异体及衍生物之类的非天然氨基酸。本发明所属技术领域的普通技术人员可以理解的是,考虑到上述广义的定义,本说明书中的氨基酸可以为例如l-氨基酸、d-氨基酸、氨基酸变异体及衍生物等化学修饰的氨基酸;正亮氨酸、β-丙氨酸、鸟氨酸等不在生物体内作为蛋白质的构成材料的氨基酸;以及本发明所属技术领域中公知的具有氨基酸特性的通过化学方法合成的化合物。非天然氨基酸的例除苏氨酸衍生
物a外,还可以有α-甲基氨基酸(α-甲基丙氨酸等)、d-氨基酸、组氨酸样氨基酸(2-氨基-组氨酸、β-羟基-组氨酸、高组氨酸、α-氟甲基-组氨酸及α-甲基-组氨酸等)、在侧链上具有多余的亚甲基的氨基酸(「同型」氨基酸)及侧链中的羧酸官能团被磺酸基取代的氨基酸(半胱氨酸等)。
56.本说明书中记载的“具有取代基的氨基酸残基”是指氨基酸残基具有取代基,可以包括例如被乙酰基取代的丙氨酸、半胱氨酸、天冬氨酸、谷氨酸、苯丙氨酸、甘氨酸、组氨酸、异亮氨酸、赖氨酸、亮氨酸、蛋氨酸、天冬酰胺、脯氨酸、谷氨酰胺、精氨酸、丝氨酸、缬氨酸、色氨酸或酪氨酸,可以为结合有氨基酸的肽。
57.本发明记载的“保护基”为在有机反应中用于保护特定官能团,例如胺基或羟基等的官能团,只要是有机合成领域中的普通技术人员认知范围内的官能团就都可以,胺保护基的具体例有芴基甲氧羰基(fmoc)、叔丁氧羰基(boc)、苄氧羰基(cbz)、二苯甲氧羰基(bhoc)、烯丙氧基羰基(alloc)、乙酰基、苯甲酰基、苄基、氨基甲酸酯、对甲氧基苄基、3,4-二甲氧基苄基、对甲氧基苯基、甲苯磺酰基、氯甲酸三氯乙酯、磺胺(bts、硝基苯磺酰基(nosyl)以及nps)、异丁酰基。
58.本发明中记载的“氯化烷烃”是指烷烃的一个以上的氢被氯取代,烷烃包括直链或支链的所有形态,除非由特别的记载,否则烷烃具有1个至10个碳原子,优选地,具有1个至7个碳原子,更优选地,具有1个至4个碳原子。
59.本说明书中记载的“取代的”、“具有取代基的”以及与取代物相关的,除非有单独的记载,否则本发明的任意取代的取代物可以使用卤素、羟基、羧酸基团、硝基、氰基、(低级)烷基、卤代烷基、单或二烷基氨基、烷氧基、硫代烷基、环烷基、杂环烷基、芳基、杂芳基、-no2、-nr
a1rb1
、-nra1c(=o)r
b1
、-nr
a1
c(=o)nr
a1rb1
、-nr
a1
c(=o)or
b1
、-nr
a1
so2r
b1
、-or
a1
、-cn、-c(=o)r
a1
、-c(=o)or
a1
、-c(=o)nr
a1rb1
、-oc(=o)r
a1
、-oc(=o)or
a1
、-oc(=o)nr
a1rb1
、-nr
a1
so2r
b1
、-po3r
a1
、-po(or
a1
)(or
b1
)、-so2r
a1
、-s(o)r
a1
、-so(nr
a1
)r
b1
(例如砜基亚胺(sulfoximine))、-s(nr
a1
)r
b1
(例如硫亚胺(sulfilimine))及-sr
a1
,其中,r
a1
与r
b1
可以相同或不同,可以各自独立地为氢、卤素、氨基、烷基、烷氧基烷基、卤代烷基、芳基和杂环,或者与附着的氮原子一样,r
a1
与r
b1
可以为杂环形态。其中,根据结合的原子,r
a1
与r
b1
可以为多个,优选地,上述烷基可以为c
1-6
烷基,环烷基及杂环烷基可以为c
3-12
,芳基可以为c
6-12
,杂环及杂芳基可以为c
3-12
。
60.本发明中记载的肽核酸单体包含肽核酸基本骨架,具体地,包含n-(2-氨基乙基)甘氨酸)(化合物5)或所有化合物8的n位点具有或不具有胺基或羟基保护基的核酸碱基。
61.具体地,还包括所有不具有羟基或不具有胺基等的下述结构。
62.本发明中记载的肽核酸二聚体由两个结合有作为肽核酸基本骨架的n-(2-氨基乙基)甘氨酸)(化合物5)或化合物8的n电位结合有核酸碱基的肽核酸单体连接而成,肽核酸二聚体所包含的核酸碱基可以互不相同或相同,具体地,可以由上述化学式11表示。
63.本发明中记载的肽核酸三聚体由三个结合有作为肽核酸基本骨架的n-(2-氨基乙
基)甘氨酸)(化合物5)或化合物8的n位结合有核酸碱基的肽核酸单体连接而成,肽核酸三聚体所包含的核酸碱基可以互不相同或相同,具体地,可以由上述化学式12表示。
64.并且,与肽核酸三聚体相同,本发明中记载的肽核酸四聚体是指由四个肽核酸单体连接而成的结构,具体地,由上述化学式13表示。
65.本说明书中记载的“肽核酸嵌段”是指两个以上的肽核酸单体结合的含义,是指肽核酸二聚体、肽核酸三聚体、肽核酸四聚体等。
66.本发明提供可以在溶液相中以高纯度及收率制备肽核酸寡聚体的方法,本发明的肽核酸寡聚体的制备方法的特征在于,包括在溶液相中使肽核酸嵌段与肽核酸嵌段反应来制备肽核酸寡聚体的步骤,上述肽核酸嵌段由不同或相同的两个以上的肽核酸单体结合而成。
67.本发明的肽核酸寡聚体的制备方法与在支撑体上一个一个连接肽核酸单体的现有的方法不同,在溶液相中制备由两个以上的肽核酸单体结合的肽核酸嵌段后,使这些肽核酸嵌段反应来制备肽核酸寡聚体,从而可以通过短的工序步骤正确地制备目标肽核酸寡聚体。
68.进而,本发明的肽核酸寡聚体的制备方法能够利用肽核酸嵌段以更高的纯度及收率制备肽核酸寡聚体,在溶液相中进行所有的反应,因此具有制备更为容易并可以大量生产的优点。
69.优选地,本发明的肽核酸寡聚体的制备方法包括在存在偶联试剂及溶剂的情况下,将由下述化学式1表示的第一肽核酸二聚体、由下述化学式2表示的第一肽核酸三聚体或由下述化学式3表示的第一肽核酸四聚体与由下述化学式4表示的第二肽核酸二聚体、由下述化学式5表示的第二肽核酸三聚体或由下述化学式6表示的第二肽核酸四聚体混合后加入胺类化合物来制备由下述化学式7至化学式9表示的肽核酸寡聚体的步骤。
70.化学式1
71.化学式2
72.化学式3
73.化学式4
74.化学式5
75.化学式6
76.化学式7
77.化学式8
78.化学式9
79.在上述化学式1至化学式9中,a1至a9及a
11
至a
14
各自独立地为包含相互不同或相同的核酸碱基的肽核酸单体,p1至p
18
各自独立地为氢,或者相互不同或相同的保护基,p1与p2、p3与p4、p5与p6、p
11
与p
12
、p
13
与p
14
、p
15
与p
16
及p
17
与p
18
各自同时为氢的情况除外,a及b各自独立地为1的整数,c及d各自独立地为0或1的整数。
80.与现有的在支撑体(树脂)上结合肽核酸单体后,将其洗涤后再结合肽核酸单体来制备肽核酸寡聚体的方法不同,本发明一实施例的肽核酸寡聚体的制备方法肽利用作为核酸嵌段肽核酸二聚体、肽核酸三聚体及肽核酸四聚体来制备,合成步骤短且非常经济,与在固相中制备的方法相比,能够以高收率及纯度制备肽核酸寡聚体。
81.进而,本发明利用肽核酸二聚体、肽核酸三聚体或肽核酸四聚体制备肽核酸寡聚体,工序步骤短且经济,在溶液相中向起始物质及偶联试剂的混合物加入作为特定化合物的胺类化合物,副产物的生成低,能够以高收率制备高纯度的肽核酸寡聚体。
82.即,与混合起始物质、偶联试剂及胺类化合物来制备肽核酸寡聚体的方法不同,本发明的肽核酸寡聚体的制备方法是向混合起始物质及偶联试剂的混合液滴加胺类化合物,划时代地降低副产物的生成,能够以高收率及纯度制备肽核酸寡聚体。
83.不仅如此,据此制备的肽核酸寡聚体的分离纯化也很容易。
84.具体地,本发明一实施例的肽核酸寡聚体的制备方法利用能够以溶液工序合成的肽核酸二聚体、肽核酸三聚体或肽核酸四聚体,在制备具有n个核酸碱基的肽核酸寡聚体时,不会制备出具有n-1个核酸碱基的肽核酸寡聚体或具有n-2个核酸碱基的肽核酸寡聚体作为副产物,很容易从其他副产物等杂志中分离出肽核酸寡聚体,因此分离出的肽核酸寡聚体的纯度很高。
85.更优选地,本发明一实施例的肽核酸寡聚体的制备方法包括:步骤a),在存在偶联试剂及溶剂的情况下向由下述化学式1-1表示的第一肽核酸二聚体、由下述化学式2-1表示的第一肽核酸三聚体或由下述化学式3-1表示的第一肽核酸四聚体加入由下述化学式4-1
表示的第二肽核酸二聚体、由下述化学式5-1表示的第二肽核酸三聚体或由下述化学式6-1表示的第二肽核酸四聚体来获得混合液;以及步骤b),向上述步骤的混合液加入胺类化合物来制备上述化学式7至化学式9的化合物。
86.化学式1-1
87.化学式2-1
88.化学式3-1
89.化学式4-1
90.化学式5-1
91.化学式6-1
92.在上述化学式1-1、化学式2-1、化学式3-1、化学式4-1、化学式5-1及化学式6-1中,a1至a9及a
11
至a
14
各自独立地为包含相互不同或相同的核酸碱基的肽核酸单体,p2、p4、p6、p7、p9及p
11
各自独立地为氢或相互不同或相同的保护基,x1至x3各自独立地为酸性盐,a及b各自独立地为1的整数,c及d各自独立地为0或1的整数。
93.优选地,为了在将化学式1-1、化学式2-1及化学式3-1的铵盐转换为游离(free)胺的同时提高与化学式4-1、化学式5-1及化学式6-1的羟基的反应性而使用作为特定胺类化合物的n,n-二异丙基乙胺,从而能够以更为提高的收率及纯度制备肽核酸寡聚体。
94.优选地,在本发明一实施例中,相对于1摩尔的上述化学式1-1、化学式2-1及化学式3-1的化合物,可以使用5摩尔至40摩尔的胺类化合物,更优选地,可以使用10摩尔至30摩尔的胺类化合物。
95.并且,本发明一实施例的肽核酸寡聚体的制备方法还可以包括再次利用上述步骤b)中获得的产物作为起始物质来重复执行步骤a)及步骤b)的步骤。
96.本发明一实施例的肽核酸寡聚体的制备方法利用肽核酸二聚体,肽核酸三聚体或肽核酸四聚体,与现有的方法不同,无需用来保护未反应的官能团的覆盖(capping)步骤,具有能够划时代地缩短制备步骤的优点,因此可以大量生产。
97.本发明一实施例的肽核酸寡聚体可以轻松制备为具有目标个数的核酸碱基的肽核酸寡聚体,但优选地,可以包含4个以上的核酸碱基,更优选地,可以包含4个至40个核酸碱基。
98.本发明一实施例的上述混合液内的起始物质与溶剂的体积比可以为1∶10以上,优选地,可以为1∶10至1∶250,更优选地,可以为1∶10至1∶100。
99.在本发明一实施例的混合液内的起始物质与溶剂的体积比为1∶10以上的情况下,能够以更高的收率及纯度制备肽核酸寡聚体。
100.本发明一实施例的核酸碱基可以为腺嘌呤、胞嘧啶、5-甲基胞嘧啶、鸟嘌呤、胸腺嘧啶、尿嘧啶、嘌呤、2,6-二氨基嘌呤、n4n
4-桥亚乙基胞嘧啶、n6n
6-桥亚乙基-2,6-二氨基嘌呤、5-(c3-c6)-炔基尿嘧啶、5-(c3-c6)-炔基-胞嘧啶、5-(1-炔丙基氨基)尿嘧啶、5-(1-炔丙基氨基)胞嘧啶、吩恶嗪、9-氨基乙氧基吩恶嗪、5-氟尿嘧啶、假异胞嘧啶、5-(羟甲基)尿嘧啶、5-氨基尿嘧啶、假尿嘧啶、二氢尿嘧啶、5-(c1-c6)-烷基尿嘧啶、5-(c1-c6)-烷基-胞嘧啶、5-(c2-c6)-烯基胞嘧啶、5-氟胞嘧啶、5-氯尿嘧啶、5-氯胞嘧啶、5-溴尿嘧啶、5-溴胞嘧啶、7-脱氮腺嘌呤、7-脱氮鸟嘌呤、8-氮杂嘌呤、7-脱氮-7-取代嘌呤,硫脲嘧啶或人造核酸碱基。
101.本发明一实施例的保护基可以为芴基甲氧羰基、叔丁氧羰基、苄氧羰基、二苯甲氧羰基、乙酰基、苯甲酰基、苄基、氨基甲酸酯、对甲氧基苄基、3,4-二甲氧基苄基、对甲氧基苯基、甲苯磺酰基、氯甲酸三氯乙酯、磺胺或异丁酰基,优选地,胺保护基可以为叔丁氧羰基、苄氧羰基、二苯甲氧羰基、乙酰基、苯甲酰基、苄基,羟基的保护基可以为(c1-c4)烷基。
102.具体地,本发明一实施例的上述第一肽核酸二聚体或第二肽核酸二聚体可以由下述化学式11表示,上述第一肽核酸三聚体或第二肽核酸三聚体可以由下述化学式12表示,上述第一肽核酸四聚体或第二肽核酸四聚体可以由下述化学式13表示。
103.化学式11
104.化学式12
105.化学式13
106.在上述化学式11至化学式13中,r1至r
18
各自独立地为氢、氨基酸的残基或具有取
代基的氨基酸残基,t1至t3各自独立地为胺保护基,z1至z3各自独立地为羟基保护基,b1至b9各自独立地为包含或不包含胺保护基核酸碱基。
107.相对于1摩尔的由上述化学式1-1表示的第一肽核酸二聚体、由上述化学式2-1表示的第一肽核酸三聚体或由上述化学式3-1表示的第一肽核酸四聚体,可以使用1摩尔∶0.8摩尔至1摩尔∶1.2摩尔的由上述化学式4-1表示的第一肽核酸二聚体、由上述化学式5-1表示的第一肽核酸三聚体或由上述化学式6-1表示的第一肽核酸四聚体优选地,可以使用1摩尔∶0.9摩尔至1摩尔∶1.1摩尔。
108.本发明一实施例的偶联试剂可以为苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(pybop,(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate)或它们的混合物,优选地,可以为苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(hbtu,n,n,n
′
,n
′‑
tetramethyl-o-(1h-benzotriazol-1-yl)uronium hexafluorophosphate),但不限定于此。
109.相对于1摩尔的由上述化学式1-1表示的第一肽核酸二聚体、由上述化学式2-1表示的第一肽核酸三聚体或由上述化学式3-1表示的第一肽核酸四聚体,可以使用1.2摩尔至2.5摩尔的偶联试剂,优选地,可以使用1.2摩尔至2.0摩尔的偶联试剂,但不限定于此。
110.本发明一实施例的步骤b)为发生偶联反应的步骤,可以在-10℃至5℃的温度下进行10分钟至60分钟,更优选地,可以在-5℃至5℃的温度下进行10分钟至40分钟。
111.本发明一实施例的溶剂可以为氯化(c1-c4)烷烃、n,n-二甲基甲酰胺及n,n-二异丙基乙胺的混合溶剂,优选地,可以为n,n-二甲基甲酰胺。
112.在此情况下,使用的溶剂可以为氯化(c1-c4)烷烃、n,n-二甲基甲酰胺(dimethylformamide)及n,n-二异丙基乙胺(diea,n,n-diisopropylethylamine)的混合溶剂,氯化(c1-c4)烷烃可以为选自三氯甲烷、二氯甲烷、氯甲烷、1,1,2-三氯乙烷、1,1,1-三氯乙烷、1,2-二氯乙烷、1,1-二氯乙烷中的一种以上。
113.本发明一实施例的溶剂使用n,n-二甲基甲酰胺、n,n-二异丙基乙胺或氯化烷烃,在偶联反应时适当调节反应物的溶解度,从而能够以高收率及纯度获得产物,从这个方面来看,优选地,溶剂可以为n,n-二甲基甲酰胺。
114.根据本发明一实施例制备的化学式7至化学式9的肽核酸寡聚体可以通过本发明所属技术领域的普通技术人员可知的多种方法分离纯化,优选地,可以通过加入水来分离纯化。
115.并且,本发明一实施例的肽核酸寡聚体的制备方法的全部工序都在溶液相中进行,与利用支撑体(树脂)来在固相中由肽核酸单体制备肽核酸寡聚体的现有的方法相比,可以通过使用肽核酸二聚体、肽核酸三聚体或肽核酸四聚体来以惊人提高的收率及纯度制备肽核酸寡聚体,是非常有效且经济的方法。
116.即,本发明的肽核酸寡聚体的制备方法可以溶液相中制备第一肽核酸二聚体、第一肽核酸三聚体、第一肽核酸四聚体、第二肽核酸二聚体,第二肽核酸三聚体、第二肽核酸四聚体及肽核酸寡聚体,以简单的工序制备具有优秀的纯度及收率的肽核酸寡聚体,具有可以大量生产的优点。
117.本发明一实施例的肽核酸寡聚体在下述条件下的高效液相色谱法(hplc)分析结果可以为70%以上的纯度,优选地,可以为75%以上的纯度,更优选地,可以为80%以上的
纯度。
118.展开溶剂:0.1%的三氟乙酸水溶液(tfa 0.1%in water),0.1%的三氟乙酸乙腈溶液(tfa0.1%in mecn),梯度条件(gradient condition)的紫外线(uv)检测仪,色谱柱250mm
×
4.6mm。
119.并且,本发明的肽核酸寡聚体的制备方法以没有覆盖过程的方式合成,制备的肽核酸寡聚体的忖度在上述条件的高效液相色谱法分析结果为70%以上,优选地,为75%以上,更优选地,为80%以上。
120.以下,通过实施例具体说明奔放买那个,但本发明的范畴不限定于下述实施例。
121.用于反映的物质、有机溶剂从novabiochem公司、alfa aesar公司、sa二氯甲烷(mc)hun chemicals公司、junsei chemicals co.,ltd公司、duksan reagents chemical公司等购入来使用,以不进行追加纯化的方式使用。合成的化合物的1h-核磁共振(1h-nmr)分析在常温下使用bruker公司的400mhz或500mhz的核磁共振谱仪进行,hplc(waters 1525binary hplc pump)将包含0.1%的三氟乙酸(tfa)的乙腈(mecn)∶包含0.1%的三氟乙酸的水的比例为5∶95的溶剂用作高效液相色谱仪(waters 1525binary hplc pump)的展开溶剂,逐渐改变展开溶剂的比例,使包含0.1%的三氟乙酸的乙腈∶包含0.1%的三氟乙酸的水的比例在20分钟内转变为20∶80,然后,在10分钟内使用包含0.1%的三氟乙酸的乙腈∶包含0.1%的三氟乙酸的水的比例为95∶5的溶剂,以柱温箱(column heater)60℃的方法进行分析。
122.以与韩国授权专利第10-0464261号相同的方法制备下述化合物1至化合物4的核酸碱基或具有胺保护基核酸碱基。
123.制备例1.制备化合物5(boc-aeg-oet)
124.向500ml的锥形瓶放入11.0g的乙二胺(ethylenediamine)(183mmol)后溶于二氯甲烷(mc)。将5.0g的boc2o(22.9mmol)溶于二氯甲烷后逐滴加入(dropwise),在常温下搅拌12小时。通过薄层色谱法(tlc)确认反应终止后,放入水只萃取二氯甲烷层并使用sat.nacl洗涤。使用na2so4处理后,过滤后浓缩。向浓缩的溶液放入二氯甲烷后加入6.4ml的三乙胺
(triethylamine)(45.8mmol)。将2.4ml的溴乙酸乙酯(ethyl bromoacetate)(22.0mmol)溶于二氯甲烷后慢慢逐滴加入,在常温下搅拌12小时。通过薄层色谱法确认反应终止后,放入水只萃取二氯甲烷层。使用na2so4去除水后,浓缩后使用硅胶色谱柱层析(sillica-column chromatoghraphy)(洗脱液:ea∶hex=1∶1(v/v))纯化来获得透明油形态产物,即,化合物5(3.79g,70%)。
125.制备例2.制备化合物8(boc-lys(z)-ome)
126.制备化合物6
127.在氮气环境下向250ml的双口(2neck)圆底烧瓶放入5.40g的boc-lys(z)-cooh(14.2mmol)后溶于100ml的干燥四氢呋喃(dry thf)。一次性放入9.21g的1,1'-羰基二咪唑(1,1'-carbonyldiimidazole)(56.8mmol)后,在常温下搅拌10分钟。当不再产生气泡时,在0℃的温度下将2.68g的nabh4(71.0mmol)溶于30ml的蒸馏水后慢慢加入后,搅拌30分钟。通过薄层色谱法确认反应终止后,浓缩溶剂。放入200ml的ea后移到分拣漏斗使用1m的hcl洗涤后,使用饱和盐水洗涤后使用硫酸钠(sodium sulfate)取出水来浓缩。使用硅胶色谱柱层析(洗脱液:ea∶hex=1∶1(v/v))纯化来获得透明黄色油形态的产物,即,化合物6(5.0g,96.2%)。
128.制备化合物7
129.在氮气环境下使用习惯(syringe)向250ml的双口圆底烧瓶放入8.71ml的草酰氯(oxayl chloride)(17.4mmol)后,放入20ml的干燥(dry)二氯甲烷。使用nacl、冰嵌段及甲醇(methanol)在5分钟之内将温度降至
–
20℃,向100ml的圆底烧瓶逐滴加入混合有2.47ml的干燥二甲基亚砜(dmso)(34.8mmol)与5ml的干燥二氯甲烷的溶液。5分钟后,将5.80g的boc-lys(cbz)-oh(15.8mmol)溶于20ml的干燥二氯甲烷后向反应液加入。15分钟后,加入15.2ml的n,n-二异丙基乙胺(87.1mmol)后搅拌5分钟,去除冰浴(ice bath)后确认薄层色谱法(只有ea,对茴香醛(ea only,p-anisaldehyde))。反应终止后使用nahco3洗涤后浓缩溶剂来获得透明油形态的化合物7(5.5g,95.3%)。
130.制备化合物8
131.参考filbert totsingan et.al.chirality,2009,21,245
–
253,以相同的方法制
备化合物8,并通过上述参考文献确认化合物8的合成。
132.向250ml的圆底烧瓶放入5.50g的boc-lys(cbz)-cho(15.1mmol)后放入50.0ml的甲醇,再放入2.84g的gly-ome(22.6mmol)。在0℃的温度下加入1.30ml的乙酸(acetic acid)(22.6mmol)和3.94ml的n,n-二异丙基乙胺(n,n-diisopropylethylamine)(22.6mmol)后,放入9.60g的三乙酰氧基硼氢化钠(sodium triacetoxyborohydride)(45.3mmol)在0℃的温度小搅拌2小时后搅拌过夜(overnight)。通过薄层色谱法(展开液:只有ea,对茴香醛)确认起始物质是否反应后,在反应终止时浓缩溶液后,放入ea并使用nahco3洗涤后浓缩,使用硅胶色谱柱层析(洗脱液:ea∶hex=1∶1(v/v))纯化来获得透明油形态的产物(4.5g,68.2%)。
133.制备例3.制备化合物11(boc-glu(ochex)-ome)
134.制备化合物9
135.向500ml的单口(1neck)圆底烧瓶放入20g的boc-glu(ochex)-cooh(60.7mmol)后溶于150ml的干燥n,n-二甲基甲酰胺(n,n-dimethylformamide)。放入22.3g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(hbtu,o-(1h-benzotriazol-1-yl-n,n,n’,n
’‑
tetramethyluroniumhexafluorophosphate))(58.3mmol)。在0℃的温度下加入23.3ml的n,n-二异丙基乙胺)(133.6mmol)后放入7.11g的n-甲氧基-n-甲胺盐酸盐(n-methoxy-n-methylamine hydrochloride)(72.9mmol),在常温下过夜搅拌。通过薄层色谱法(展开液:3%的乙酸(acoh),5%的甲醇(meoh)/二氯甲烷,茚三酮(ninhydrin))确认起始物质是否反应后,在反应终止时浓缩溶液后放入150ml的乙酸乙酯(ethyl acetate,ea)后,使用0.5n的hcl、饱和nahco3及1n的khso4洗涤。向有机层加入硫酸钠去除有机层的水后,浓缩来获得透明无色油形态的产物,即,化合物9(21g,92.9%)。
136.制备化合物10
137.在氮气环境下向1l的三口(3neck)圆底烧瓶放入21.0g的化合物9(56.4mmol)后溶于210ml的干燥四氢呋喃。在-78℃的温度下,在20分钟内加入26.2g的2.37m的氢化铝锂(litium aluminium hydride)(62.0mmol)后搅拌1小时。通过薄层色谱法(展开液:ea∶hex=1∶1,茚三酮)确认起始物质是否反应后,在反应终止时放入600ml的乙酸乙酯,在10分钟内加入210ml的1m的khso4后搅拌10分钟。使用1m的khso4、饱和nahco3洗涤有机层,使用硫酸钠去除水后浓缩来获得透明黄色油形态的产物,即,化合物10(18.5g,88.1%)。
138.制备化合物11
139.向500ml的圆底烧瓶放入18.5g的化合物10(59.0mmol)后,放入130ml的二氯乙烷(dichloroethane),加入甘氨酸甲酯盐酸盐(glycine methyl ester hydrochloride,hcl.gly-ome)。在0℃的温度下加入3.38ml的乙酸(59.0mmol)与15.4ml的n,n-二异丙基乙胺(88.5mmol)后,加入18.9g的三乙酰氧基硼氢化钠(88.5mmol)搅拌30分钟。通过薄层色谱法(洗脱液:ea∶hex=1∶4,洗脱两次,茚三酮)确认起始物质是否反应后,在反应终止时在0℃的温度下放入130ml的饱和nahco3后搅拌10分钟。使用饱和nahco3、饱和盐水洗涤有机层,使用硫酸钠去除水后浓缩,通过硅胶色谱柱层析(洗脱液:ea∶hex=1∶1(v/v))纯化来获得透明油形态的产物,即,化合物11(boc-glu(ochex)-ome)(14g,61.4%)。
140.制备例4.制备化合物12(boc-lys(z)-nh2)
141.制备化合物12
142.向1l的圆底烧瓶放入20.0g的boc-lys(cbz)-cooh(52.6mmol)后溶于200ml的n,n-二甲基甲酰胺。放入29.9g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(78.9mmol),放入5.62g的nh4cl(105.1mmol)后,在0℃的温度下滴加36.6ml的n,n-二异丙基乙胺(210.3mmol)。搅拌约30分钟后,利用薄层色谱法确认反应终止后加入二氯甲烷(dichloromethane)(200ml),利用分拣漏斗分别使用5%的nahco3、0.5m的hcl各洗涤一次。随着有机层慢慢变浑浊生成固体(solid),过滤(filter)来获得化合物12(boc-lys(z)-nh2)(19.9g,99.9%)。
143.实施例1.制备肽核酸单体
144.制备化合物13-3(boc-aeg-a(z)-oet)
145.向3l的圆底烧瓶放入20.0g的化合物3(61.1mmol)后溶于1l的n,n-二甲基甲酰胺。放入34.8g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(122mmol),放入15.1g的化合物
5(61.1mmol)后在0℃的温度下滴加31.9ml的n,n-二异丙基乙胺(183mmol)后搅拌30分钟。利用薄层色谱法(展开液:5%的甲醇/二氯甲烷)确认反应终止后,放入1l的二氯甲烷(dichloromethane,mc)。使用饱和nahco3、0.5m的hcl洗涤有机层后,放入硫酸钠去除水来浓缩。通过硅胶色谱柱层析(洗脱液:5%的甲醇/二氯甲烷)纯化来获得白色固体产物,即,化合物13(boc-a(z)-oet)(30.0g,88.4%)。
146.制备化合物14-3(boc-aeg-a(z)-oh)
147.向500ml的圆底烧瓶放入15.0g的化合物13-3(27.0mmol),溶于270ml的0.5n的lioh(135mmol)后搅拌约39分钟后利用薄层色谱法(洗脱液:5%的甲醇/二氯甲烷)确认反应终止。在0℃的温度下使用0.5m的hcl使ph降至2~3后,使用玻璃过滤器(glass filter)过滤生成的固体,使用水洗涤数次后使用p2o5干燥来获得白色固体形态的产物,即,化合物14-3(boc-a(z)-oh)(14.1g,99.0%)。
148.制备化合物15-1(nh
2-aeg-a(z)-oet)
149.向250ml的圆底烧瓶放入15.0g的化合物13-3(27.0mmol),在0℃的温度下溶于135ml的50%的三氟乙酸/二氯甲烷后简报30分钟。利用薄层色谱法(展开液:5%的甲醇/二氯甲烷)确认反应终止。向过量的二乙醚(diethyl ether)滴加溶液来产生沉淀后,使用玻璃过滤器过滤,使用二乙醚洗涤数次来获得白色固体形态的产物,即,化合物15-1(nh
2-a(z)-oet)(14.5g,94.3%)。
150.实施例2.制备肽核酸单体
151.除在实施例1的制备化合物13-3的过程中使用化合物1替代化合物3以外,以与实施例1相同的方法制备肽核酸单体14-1(boc-aeg-t-oh)及肽核酸单体15-1(nh
2-aeg-t-oet)。
152.实施例3.制备肽核酸单体
153.除在实施例1的制备化合物13-3的过程中分别使用化合物2或化合物4替代化合物3以外,以与实施例1相同的方法制备肽核酸单体14-2(boc-aeg-c(z)-oh)、肽核酸单体14-4(boc-aeg-g(z)-oh)及肽核酸单体15-4(nh
2-aeg-g(z)-oet)。
154.实施例4.制备肽核酸单体
155.除在实施例1的制备化合物13-3的过程中使用化合物8替代化合物5,使用化合物2替代化合物3以外,以与实施例1相同的方法制备肽核酸单体14-5(boc-lys(z)-c(z)-oh)及肽核酸单体15-6(nh
2-lys(z)-nh2)。
156.实施例5.制备肽核酸单体
157.除在实施例1的制备化合物13-3的过程中使用化合物11替代化合物5,使用化合物1替代化合物3以外,以与实施例1相同的方法制备肽核酸单体15-3(nh
2-glu(ochex)-t-ome)。
158.实施例6.制备肽核酸单体
159.除分别使用化合物2或化合物4替代化合物1以外,以与实施例5相同的方法分别制备肽核酸单体15-4(nh
2-glu(ochex)-c(z)-ome)、肽核酸单体15-5(nh
2-glu(ochex)-g(z)-ome)。
160.实施例7.制备肽核酸二聚体17-1(boc-aa-oh)
161.制备化合物16-1(boc-aa-oet)
162.向2l的圆底烧瓶放入13.0g的化合物14-3(24.6mmol)溶于650ml的干燥n,n-二甲基甲酰胺后,放入14.0g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(37.0mmol)。放入14.0g的化合物15-1(24.6mmol)后在0℃的温度下滴加21.5ml的n,n-二异丙基乙胺(123mmol)搅拌30分钟。通过薄层色谱法(洗脱液:15%的甲醇/二氯甲烷)确认反应终止后,放入650ml的二氯甲烷。使用饱和nahco3、0.5m的hcl洗涤有机层后,加入硫酸钠去除水来浓缩。通过硅胶色谱柱层析(洗脱液:10%的甲醇/二氯甲烷)纯化来获得白色固体产物boc-aa-oet(21.0g,87.9%)。
163.制备化合物17-1(boc-aa-oh)
164.向500ml的圆底烧瓶加入21.0g的化合物16-1(21.7mmol),在0℃的温度下溶于217ml的0.5n的lioh(108mmol)搅拌30分钟。通过薄层色谱法(展开液:10%的甲醇/二氯甲烷)监控来终止反应。在0℃的温度下使用1m的hcl使ph降至2~3后使用玻璃过滤器过滤生成的固体,使用水洗涤数次后使用甲醇/乙醚(ether)重结晶来获得白色的产物,即,化合物
17-1(boc-aa-oh)(19.7g,96.6%)。
165.实施例8.制备肽核酸二聚体
166.除分别使用不同的肽核酸单体替代化合物14-3及化合物15-3以外,以与实施例7相同的方法分别制备肽核酸二聚体16-2(boc-c"a-oet)、肽核酸二聚体17-2(boc-cc^-oh)、肽核酸二聚体17-3(boc-tt^-oh)及肽核酸二聚体17-4(boc-gg-oh)。在此情况下,base"为nh
2-γlys(z)-base-ome,base^为nh
2-γglu(ochex)-base-ome。
167.实施例9.制备肽核酸二聚体18-1(nh
2-c"a-oet)
168.向250ml的圆底烧瓶放入20.0g的化合物16-2(boc-c"a-oet)(17.4mmol),在0℃的温度下溶于180ml的50%的三氟乙酸/二氯甲烷后搅拌30分钟。利用薄层色谱法(洗脱液:7%的甲醇/二氯甲烷)终止反应。向过量的二乙醚滴加溶液来产生沉淀后,使用玻璃过滤器过滤后使用二乙醚洗涤来获得白色固体产物nh
2-c"a-oet(19.2g,94.9%)。
169.实施例10.制备ok linker(化合物18-2,nh
2-ok-nh2)
170.制备化合物16-6(boc-ok-nh2)
171.向2l的圆底烧瓶放入13.0g的3,6,11-三恶-9-氮杂十三烷酸,12,12-二甲基-氧代-97%(boc-o-oh)(3,6,11-trioxa-9-azatridecanoic acid,12,12-dimethyl-10-oxo-97%(boc-o-oh))(49.4mmol)溶于650ml的干燥n,n-二甲基甲酰胺后,放入22.5g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(59.3mmol)。放入19.4g的化合物15-6(49.4mmol)后在0℃的温度下滴加21.5ml的n,n-二异丙基乙胺(123mmol)后搅拌30分钟。利用薄层色谱法(展开液:7%的甲醇/乙酸乙酯)确认反应终止后,放入650ml的二氯甲烷。使用饱和nahco3、0.5m的hcl洗涤有机层后,加入硫酸钠去除水来浓缩。通过硅胶色谱柱层析(洗脱液:7%的甲醇/乙酸乙酯)纯化来获得透明无色油形态的boc-ok-nh2(21.0g,81.1%)。
172.制备化合物18-2(nh
2-ok-nh2)
173.向250ml的圆底烧瓶加入21.0g的化合物16-6(boc-ok-nh2)(40.0mmol)在0℃的温
度下溶于180ml的50%的三氟乙酸/二氯甲烷后搅拌30分钟。利用薄层色谱法(展开液:7%的甲醇/乙酸乙酯)终止反应。向过量的二乙醚滴加溶液产生沉淀后,将通过离心分离(centrifuge)获得的油形态的产物溶于二氯甲烷后浓缩溶剂来获得透明油形态的产物nh
2-ok-nh2(19.2g,89.1%)。
174.实施例11.制备肽核酸三聚体19-1(boc-aag^-ome)
175.向2l的圆底烧瓶放入15.2g的化合物17-1(16.2mmol)溶于750ml的干燥的n,n-二甲基甲酰胺。加入12.6g的六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(pybop,benzotriazole-1-yl-oxytripyrrolidinophosphate)后,放入11.7g的化合物15-5。在0℃的温度下放入56.3ml的n,n-二异丙基乙胺搅拌约30分钟后,利用薄层色谱法(展开液:20%的甲醇/二氯甲烷)确认反应终止。放入300ml的二氯甲烷后使用饱和nahco3、0.5m的hcl溶液洗涤有机层,使用硫酸钠去除水来浓缩后获得透明的黄色油形态的产物。使用甲醇/乙醚使该产物重结晶来获得白色固体化合物,即,化合物19-1(boc-aag^-ome)(21.3g,85.9%)。
176.实施例12.制备肽核酸三聚体19-2(boc-cc"a-oet)
177.除使用肽核酸单体14-2、肽核酸二聚体18-1替代肽核酸二聚体17-1及肽核酸单体15-5以外,以与实施例11相同的方法制备肽核酸三聚体19-2(boc-cc"a-oet)。在此情况下base"为nh
2-γlys(z)-base-ome。
178.实施例13.制备肽核酸三聚体20-1(boc-aag^-oh)
179.向1l的圆底烧瓶放入20.3g的化合物19-1(13.2mmol)溶于100ml的1,4-二恶烷(1,4-dioxane)。在0℃的温度下滴加132ml的0.5m的lioh(66.1mmol)后搅拌约30分钟。利用薄层色谱法(洗脱液:20%的甲醇/二氯甲烷)确认反应终止。在0℃的温度下使用0.5m的hcl使ph降至2~3,使用玻璃过滤器过滤生成的固体,使用水洗涤后使用甲醇/乙醚使其重结晶来
获得白色固体产物,即,化合物20-1(boc-aag^-oh)(18.3g,90.8%)。
180.实施例14.制备肽核酸三聚体21-1(nh
2-cc"a-oet)
181.向250ml的圆底烧瓶加入25.3g的化合物19-2(16.5mmol)在0℃的温度下溶于230ml的50%的三氟乙酸/二氯甲烷后搅拌30分钟,利用薄层色谱法(展开液:20%的甲醇/二氯甲烷)确认反应终止。向过量的二乙醚滴加溶液生成沉淀后,使用玻璃过滤器过滤后,使用二乙醚洗涤来生成白色固体产物,即,化合物21-1(nh
2-cc"a-oet)(23.8g,93.2%)。
182.实施例15.制备肽核酸23-1(nh-2-ggok-nh2)
183.制备化合物22-1(boc-ggok-nh2)
184.向1l的圆底烧瓶放入13.8g的化合物17-4(14.2mmol)溶于650ml的n,n-二甲基甲酰胺。加入11.1g的六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(21.4mmol)后,放入7.67g的化合物18-2(14.2mmol)。在0℃的温度下放入49.6ml的n,n-二异丙基乙胺(285mmol)搅拌约30分钟后,利用薄层色谱法(洗脱液:20%的甲醇/二氯甲烷)确认反应终止。放入650ml的二氯甲烷后使用饱和nahco3、0.5m的hcl溶液洗涤有机层,使用硫酸钠去除水来浓缩后获得透明无色油形态的产物,使用甲醇/乙醚使该产物重结晶来获得白色固体化合物,即,化合物22-1(boc-ggok-nh2)(17.7g,90.4%)。
185.制备化合物23-1(nh
2-ggok-nh2)
186.向250ml的圆底烧瓶放入16.7g的化合物22-1(12.1mmol)在0℃的温度下溶于140ml的50%的三氟乙酸/二氯甲烷后搅拌30分钟后,利用薄层色谱法(展开液:20%甲醇/二氯甲烷)终止反应。向过量的二乙醚滴加溶液产生沉淀后,使用玻璃过滤器过滤,使用二乙醚洗涤来获得白色固体产物,即,化合物23-1(nh
2-ggok-nh2)(16.0g,94.8%)。
187.实施例16.制备肽核酸五聚体25-1(boc-cc^cc"a-oh)
188.制备化合物24-1(boc-cc^cc"a-oet)
189.向1l的圆底烧瓶加入6.13g的化合物17-2(5.88mmol)溶于300ml的干燥n,n-二甲基甲酰胺。放入4.59g的六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(8.82mmol)后,放入9.08g的化合物21-1(5.88mmol)后在0℃的温度下滴加20.5ml的n,n-二异丙基乙胺。搅拌约30分钟后,利用薄层色谱法(展开液:5%的乙酸,10%的甲醇/二氯甲烷)确认反应终止后,放入300ml的二氯甲烷后使用饱和nahco3、0.5m的hcl洗涤有机层。使用硫酸钠去除有机层
的水后浓缩来获得产物,使用甲醇/乙醚使该产物重结晶来获得白色固体产物,即,化合物24-1(boc-cc^cc"a-oet)(13.2g,91.4%)。
190.制备化合物25-1(boc-cc^cc"a-oh)
191.向500ml的圆底烧瓶放入13.0g的化合物24-1(5.29mmol)溶于71.5ml的1,4-二恶烷。在0℃的温度下放入52.9ml的0.5n的lioh(26.5mmol)后搅拌约30分钟。利用薄层色谱法(洗脱液:5%的乙酸,20%的甲醇/二氯甲烷)确认反应终止后,使用0.5m的hcl使ph降至2~3产生固体,使用过玻璃过滤器过滤并使用水洗涤后,使用甲醇/乙醚进行重结晶。使用玻璃过滤器过滤生成的固体后使用二乙醚洗涤后干燥来获得白色固体形态的产物,即,化合物25-1(boc-cc^cc"a-oh)(12.1g,94.2%)。
192.实施例17.制备肽核酸六聚体27-1(boc-aag^cc"a-oh)
193.制备化合物26-1(boc-aag^cc"a-oet)
194.向2l的圆底烧瓶放入11.7g的化合物20-1(7.66mmol)溶于550ml的干燥的n,n-二
甲基甲酰胺。加入4.36g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(11.5mmol)后,放入11.8g的化合物21-1(7.66mmol)。在0℃的温度下放入26.7ml的n,n-二异丙基乙胺(153mmol)后搅拌30分钟。利用薄层色谱法(展开液:5%的乙酸,15%的甲醇/二氯甲烷)确认反应终止后,放入550ml的二氯甲烷后使用饱和nahco3、0.5m的hcl洗涤有机层,使用硫酸钠去除水后浓缩来获得透明黄色油形态的产物。使用甲醇/乙醚使该化合物重结晶来获得固体化合物,即,化合物26-1(boc-aag^cc"a-oet)(收率(yield):18.2g,81.0%)。
195.制备化合物27-1(boc-aag^cc"a-oh)
196.向500ml的圆底烧瓶放入18.2g的(6.20mmol)后溶于182ml的1,4-二恶烷。在0℃的温度下滴加62.0ml的0.5n的lioh后搅拌30分钟。通过薄层色谱法(展开液:5%的乙酸,15%的甲醇/二氯甲烷)监控来终止反应。在0℃的温度下使用1m的hcl使ph降至2~3来生成固体,过滤后使用水洗涤后,使用甲醇/乙醚使干燥的固体重结晶来获得白色化合物,即,化合物27-1(15.1g,83.8%)(进行了prep纯化)。
197.实施例18.制备肽核酸四聚体衔接物化合物28-1(tfa.nh
2-tt^ggok-nh2)
198.制备化合物26-2(boc-tt^ggok-nh2)
199.除使用化合物17-3、化合物23-1替代肽核酸三聚体20-1及肽核酸三聚体21-1以外,以与实施例17相同的方法制备化合物26-2(boc-tt^ggok-nh2),在此情况下,base^为nh
2-γglu(ochex)-base-ome。
200.制备化合物28-1(tfa.nh
2-tt^ggok-nh2)
201.向250ml的圆底烧瓶放入15.5g的化合物26-2(7.52mmol)在0℃的温度下溶于140ml的50%的三氟乙酸/二氯甲烷后搅拌30分钟。通过薄层色谱法(展开液:20%的甲醇/二氯甲烷)监控来终止反应。反应终止后,在0℃的温度下过量的二乙醚滴加溶液来产生沉淀。过滤后获得白色固体形态的产物,即,化合物28-1(tfa.nh
2-tt^ggok-nh2)(14.6g,94.3%)。
202.实施例19.制备肽核酸十聚体衔接物化合物30-1(nh
2-cc^cc"aatt^ggok-nh2)
203.制备化合物29-1(boc-cc^cc"att^ggok-nh2)
204.向1l的圆底烧瓶放入5.76g的化合物25-1(2.37mmol)溶于250ml的干燥的n,n-二甲基甲酰胺后,放入1.85g的六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(3.56mmol)。放入4.92g的化合物28-1(2.37mmol)后在0℃的温度下滴加12.0ml的n,n-二异丙基乙胺(68.8mmol)后搅拌30分钟。使用高效液相色谱仪(hplc)(agilent 1100型,在20分钟内使乙腈从10%到70%,之后在25分钟内到达100%(10%mecn 70%for 20mins 100%for 25mins))来检测制备的粗(crude)肽核酸寡聚体的反应终止和粗纯度(crude purity)。放入250ml的二氯甲烷后使用饱和nahco3、0.5m的hcl洗涤有机层后,使用硫酸钠去除水后浓缩来获得透明黄色油形态的产物,使用甲醇/乙醚使该产物重结晶来获得白色固体形态的化合物29-1(boc-cc^cc"att^ggok-nh2)(8.32g,80.3%)。
205.制备化合物30-1(tfa.nh
2-cc^c c"att^ggok-nh2)
206.向250ml的圆底烧瓶放入8.32g的化合物30-1(1.90mmol)在0℃的温度下溶于140ml的50%的三氟乙酸/二氯甲烷后搅拌30分钟。使用高效液相色谱仪(agilent 1100型,在20分钟内使乙腈从10%到70%,之后在25分钟内到达100%)来检测制备的粗肽核酸寡聚体的反应终止和粗纯度。反应终止后,在0℃的温度下向过量的二乙醚滴加溶液来产生沉淀。过滤后获得白色固体形态的产物,即,化合物30-1(tfa.nh
2-cc^cc"att^ggok-nh2)(7.31g,87.6%)(进行了prep纯化)。
207.实施例20.制备肽核酸十五聚体31-1(boc-aag^cc"acc^cc"att^ggok-nh2)
208.向500ml的圆底烧瓶放入3.54g的化合物20-1(1.22mmol)溶于350ml的干燥的n,n-二甲基甲酰胺后放入0.693g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(1.83mmol)。放入5.34g的化合物30-1(1.22mmol)后,在0℃的温度下放入4.24ml的n,n-二异丙基乙胺
(24.4mmol)后搅拌30分钟。使用高效液相色谱仪(agilent 1100型,在20分钟内使乙腈从10%到70%,之后在25分钟内到达100%)来测量制备的粗肽核酸寡聚体的粗纯度,结果如图1所示。
209.如图1所示,测量的粗纯度87%。
210.比较例1.制备化合物31-1(boc-aag^cc"acc^cc"att^ggok-nh2)
211.向500ml的圆底烧瓶放入1.00g的化合物20-1(0.344mmol)溶于50ml的干燥的n,n-二甲基甲酰胺后加入0.196g的苯并三氮唑-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.516mmol),在常温下搅拌0.6ml的n,n-二异丙基乙胺(3.44mmol)10分钟。将1.51g的化合物30-1(0.344mmol)溶于50ml的干燥的n,n-二甲基甲酰胺后放入0.6ml的n,n-二异丙基乙胺(3.44mmol)在0℃的温度下搅拌10分钟。在0℃的温度下混合制备的溶液并搅拌30分钟后,使用高效液相色谱仪(agilent 1100型,在20分钟内使乙腈从10%到70%,之后在25分钟内到达100%)来测量制备的粗肽核酸寡聚体的粗纯度,结果如图2所示。
212.如图2所示,可知粗纯度随着添加胺类化合物的顺序的不同而有明显的差异。
213.实施例21.制备肽核酸寡聚体
214.除使用30ml的干燥的n,n-二甲基甲酰胺以外,以与实施例20相同的方法制备肽核酸寡聚体31-1(boc-aag^cc"acc^cc"att^ggok-nh2)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。