1.本发明属于高分子材料制备应用领域,具体涉及一种超高分子量聚氯烯烃共聚物实心微球及其制备方法。
背景技术:
2.聚氯烯烃分子结构中的氯原子使其具有优越的化学性质,如:优异的化学惰性,高度稳定性,良好的绝缘性和耐候性,及高透明度、硬度、刚性等。其中,聚三氟氯乙烯的优异性能使它不仅应用于医用、化工领域,同样在尖端技术和军事宇航领域、电子工业领域具有不可替代的地位。
3.目前,所使用的聚合方法,仅能得到分子量在10~20万之间的聚三氟氯乙烯,低分子量严重影响着聚合物的机械性能,包括:抗摩擦性,抗拉伸性,抗冲击性等。聚氯烯烃的合成方式包括溶液聚合,悬浮聚合和乳液聚合。工业上常用的为悬浮聚合和乳液聚合:乳液聚合聚合速率快,可以得到较高分子量的聚氯烯烃共聚物,但后处理复杂,生产成本较高,并且难以除尽乳化剂,有损聚合物性能。传统的悬浮聚合后处理简单,生产成本较低,但生产周期较长,生产效率较低,所得聚合物分子量较低。而在一定范围内,聚合物的溶液粘度以及机械强度等性质随其分子量增加而增大,制备超高分子量聚氯烯烃共聚物,可以满足聚合物高性能要求。
技术实现要素:
4.针对上述问题情况,本发明提供一种超高分子量聚氯烯烃共聚物实心微球及其制备方法,解决现有工艺制备的聚氯烯烃共聚物分子量偏低的技术问题。
5.为了实现上述目的,本发明采取的技术方案为:一种超高分子量聚氯烯烃共聚物实心微球的制备方法,包括如下步骤:(1)对反应釜进行抽真空后充入氮气,在氮气气氛下,按照一定原料质量比加入第二单体、催化剂、引发剂、缓冲剂、分散剂以及去离子水,关闭反应釜;(2)将反应釜升温至20~100℃,维持搅拌转速500~1500 r/min,充入聚氯烯烃单体,达到聚合反应压力;(3)当反应压力下降时,补充聚氯烯烃单体,维持反应压力,维持反应温度20~100℃,反应5~15 h;(4)反应结束后对聚合产物进行离心、洗涤、干燥,得到超高分子量聚氯烯烃共聚物实心微球。
6.作为进一步的技术方案,步骤(1)中原料质量比为:第二单体:催化剂:引发剂:缓冲剂:分散剂:去离子水= 5-20: 0.0001-0.01: 0.1-1.0: 0.1-1.0: 0.1-1.0: 300-800。
7.作为进一步的技术方案,所述步骤(1)中第二单体为甲基丙烯酸六氟丁酯,偏氟乙烯,全氟己烯,全氟庚烯中的一种或几种,第二单体的主要作用是改性,增加产物的柔性和分子量,提高产物的机械性能,步骤(2)中氯烯烃为三氟氯乙烯,氯乙烯,氯丙烯中的一种,
步骤(2)中三氟氯乙烯的反应压力是0.2~2.0 mpa。
8.作为进一步的技术方案,步骤(1)中催化剂为金属纳米颗粒,所述金属纳米颗粒为金纳米颗粒,银纳米颗粒,铂纳米颗粒,铜纳米颗粒,镍纳米颗粒,铁纳米颗粒,钯纳米颗粒,钌纳米颗粒中的一种。
9.作为进一步的技术方案,金属纳米颗粒为球状,柱状,线状,六面体状,三角形状,哑铃状,球状金属纳米颗粒的直径为2~100 nm。
10.作为进一步的技术方案,步骤(1)中引发剂为2-溴异丁酸乙酯或2-溴丙酸乙酯;步骤(1)中缓冲剂为磷酸二氢钠、磷酸二氢钾、四硼酸钠、柠檬酸钠、碳酸钠或醋酸钠中的一种或一种以上;步骤(1)中分散剂为聚乙烯醇。
11.本发明的一种超高分子量聚氯烯烃共聚物实心微球的制备方法,采用金属纳米颗粒催化聚合反应体系,聚合方式为悬浮聚合。
12.本发明还提出了一种上述制备方法制得的超高分子量聚氯烯烃共聚物实心微球。
13.作为进一步的技术方案,所述超高分子量聚氯烯烃共聚物实心微球的分子量是普通聚合所得聚合物的1.5~3.0倍,分子量分布为2.5~5.2,微球尺寸为10~200
ꢀµ
m。
14.本发明与现有技术相比具有的有益效果为:本发明的一种超高分子量聚氯烯烃共聚物实心微球及其制备方法,通过金属纳米颗粒催化的新型聚合反应体系,卤代烃引发,使用悬浮聚合的方式,合成超高分子量聚氯烯烃共聚物实心微球,所用方法,成本可控,对环境友好,所得聚合物分子量是普通聚合所得聚合物的1.5~3.0倍的超高分子量,机械性能优良,满足高性能聚氯烯烃共聚物的要求。以最优的配比进行反应,可以得到分子量大于40万的超高分子量聚氯烯烃共聚物实心微球。
15.附图说明
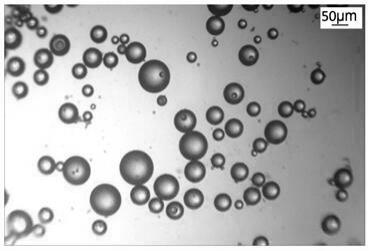
16.图1为本发明实施例 1 制备的超高分子量聚氯烯烃共聚物实心微球的外貌图;图2为本发明实施例 1 制备的超高分子量聚氯烯烃共聚物实心微球的gpc流出曲线图。
17.具体实施方式
18.为使本发明的上述目的、特征和优点能够更为明显易懂,下面对本发明的具体实施例作详细说明。
19.实施例1:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入600 g去离子水,10 g甲基丙烯酸六氟丁酯,0.001 g银纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.3 g柠檬酸钠,搅拌速率控制在800 r/min,充分搅拌。
20.升温至40℃,充入三氟氯乙烯使釜压升至1.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力1.0 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
21.微球的粒径测量:取干燥后的聚三氟氯乙烯实心微球用扫描电子显微镜(sem)进行观察。
22.分子量测试:取一定量产物,溶解于四氢呋喃溶剂,通过凝胶渗透色谱法(gpc)测出聚合物色谱图,分析计算出聚合物实心微球的分子量。
23.所得聚三氟氯乙烯共聚物实心微球分子量为41万,分子量分布为3.5,微球尺寸50~140
ꢀµ
m,产物收率=93.2%。
24.实施例2:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入800 g去离子水,5 g甲基丙烯酸六氟丁酯,0.0001 g银纳米颗粒,0.1 g 2-溴异丁酸乙酯,0.1 g 聚乙烯醇,0.1 g柠檬酸钠,搅拌速率控制在500 r/min,充分搅拌。
25.升温至20℃,充入三氟氯乙烯使釜压升至0.2 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力0.2 mpa,聚合温度保持20℃,聚合反应时间为15 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
26.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为30.8万,分子量分布为2.5,微球尺寸10~110
ꢀµ
m,产物收率=96.3%。
27.实施例3:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入300 g去离子水,20 g甲基丙烯酸六氟丁酯,0.01 g银纳米颗粒,1.0 g 2-溴异丁酸乙酯,1.0 g 聚乙烯醇,1.0 g磷酸二氢钠,搅拌速率控制在1500 r/min,充分搅拌。
28.升温至100℃,充入三氟氯乙烯使釜压升至2.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力2.0 mpa,聚合温度保持100℃,聚合反应时间为5 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
29.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为36.2万,分子量分布为5.0,微球尺寸50~110
ꢀµ
m,产物收率=98.5%。
30.实施例4:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入600 g去离子水,10 g甲基丙烯酸六氟丁酯,0.001 g铜纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.3 g磷酸二氢钾,搅拌速率控制在800 r/min,充分搅拌。
31.升温至40℃,充入三氟氯乙烯使釜压升至1.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力1.0 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
32.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为35.5万,分子量分布为3.8,微球尺寸30~110
ꢀµ
m,产物收率=88.9%。
33.实施例5:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入600 g去离子水,10 g甲基丙烯酸六氟丁酯,
0.001 g镍纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.3 g磷酸二氢钠,搅拌速率控制在800 r/min,充分搅拌。
34.升温至40℃,充入三氟氯乙烯使釜压升至1.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力1.0 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
35.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为45.0万,分子量分布为4.3,微球尺寸40~115
ꢀµ
m,产物收率=95.8%。
36.实施例6:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入600 g去离子水,10 g全氟己烯,0.001 g钯纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.3 g磷酸二氢钠,搅拌速率控制在800 r/min,充分搅拌。
37.升温至40℃,充入三氟氯乙烯使釜压升至1.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力1.0 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
38.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为35.5万,分子量分布为3.9,微球尺寸20~100
ꢀµ
m,产物收率86.7%。
39.实施例7:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置加入600 g去离子水,10 g全氟庚烯,0.001 g银纳米颗粒,0.8 g 2-溴丙酸乙酯,0.3 g 聚乙烯醇,0.3 g四硼酸钠,搅拌速率控制在800 r/min,充分搅拌。
40.升温至40℃,充入三氟氯乙烯使釜压升至1.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力1.0 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
41.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为46.8万,分子量分布为4.6,微球尺寸30~120
ꢀµ
m,产物收率=95.0%。
42.实施例8:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置600 g去离子水,10 g甲基丙烯酸六氟丁酯,0.001 g铂纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.2 g柠檬酸钠,0.1g醋酸钠,搅拌速率控制在1000 r/min,充分搅拌。
43.升温至40℃,充入三氟氯乙烯使釜压升至1.0 mpa,压力下降时补加三氟氯乙烯单体,维持反应压力1.0 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚三氟氯乙烯共聚物实心微球。
44.重复实施例一中步骤。所得聚三氟氯乙烯共聚物实心微球分子量为38.8万,分子量分布为3.5,微球尺寸40~200
ꢀµ
m,产物收率=90.5%。
45.实施例9:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈
钢高压釜内,氮气气氛下,通过加料装置600 g去离子水,10 g甲基丙烯酸六氟丁酯,0.001 g铂纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.2 g柠檬酸钠,0.1g醋酸钠,搅拌速率控制在1000 r/min,充分搅拌。
46.升温至40℃,充入氯乙烯使釜压升至0.2 mpa,压力下降时补加氯乙烯单体,维持反应压力0.2 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚氯乙烯共聚物实心微球。
47.重复实施例一中步骤。所得聚氯乙烯共聚物实心微球分子量为30.5万,分子量分布为3.2,微球尺寸40~200
ꢀµ
m,产物收率=90.5%。
48.实施例10:密闭反应釜,对反应釜进行抽真空,然后向反应釜中充入氮气。在配有搅拌的不锈钢高压釜内,氮气气氛下,通过加料装置600 g去离子水,10 g甲基丙烯酸六氟丁酯,0.001 g铂纳米颗粒,0.8 g 2-溴异丁酸乙酯,0.3 g 聚乙烯醇,0.2 g柠檬酸钠,0.1g醋酸钠,搅拌速率控制在1000 r/min,充分搅拌。
49.升温至40℃,充入氯丙烯使釜压升至0.2 mpa,压力下降时补加氯乙烯单体,维持反应压力0.2 mpa,聚合温度保持40℃,聚合反应时间为10 h。反应结束后,停止搅拌,对聚合产物进行离心、洗涤、干燥,得到超高分子量聚氯丙烯共聚物实心微球。
50.重复实施例一中步骤。所得聚氯丙烯共聚物实心微球分子量为32.5万,分子量分布为3.8,微球尺寸40~200
ꢀµ
m,产物收率=88.5%。
51.需要说明的是,本发明工艺中的含氯烯烃不限于上述含氯单体,可以为其他含氯烯烃,同样适用于本发明的工艺方法,制得聚氯烯烃共聚物实心微球。
52.本发明的一种超高分子量聚氯烯烃共聚物实心微球及其制备方法,通过金属纳米颗粒催化的新型聚合反应体系,卤代烃引发,使用悬浮聚合的方式,合成超高分子量聚氯烯烃共聚物实心微球,微球尺寸在10~200
ꢀµ
m。所得产物分子量高,稳定性及机械性能优越。所用方法,成本可控,对环境友好,所得聚合物具有普通聚合所得聚合物的1.5~3.0倍的超高分子量,机械性能优良,满足高性能聚氯烯烃共聚物的要求。以最优的配比进行反应,可以得到分子量大于40万的超高分子量聚氯烯烃共聚物实心微球,产物收率在87%~99%之间,收率高。
53.虽然本发明披露如上,但本发明并非限定于此。任何本领域技术人员,在不脱离本发明的精神和范围内,均可作各种更动与修改,因此本发明的保护范围应当以权利要求所限定的范围为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
