2005,70,14,5508;tetrahedron asymmetry2011,22,1855)。
[0008][0009]
综合来说,目前合成(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的方法存在着一定的局限性,工业化大批量生产安全风险较高。
技术实现要素:
[0010]
现有的(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的合成方法不适合工业化放大生产,生产存在安全风险的问题。针对上述的技术问题,本发明的目的在于提供一种(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯新的合成方法,该合成方法原料易得,不存在高危反应,工艺叠缩,收率高,放大生产效果好,适合于大规模工业生产。具体技术方案如下:
[0011]
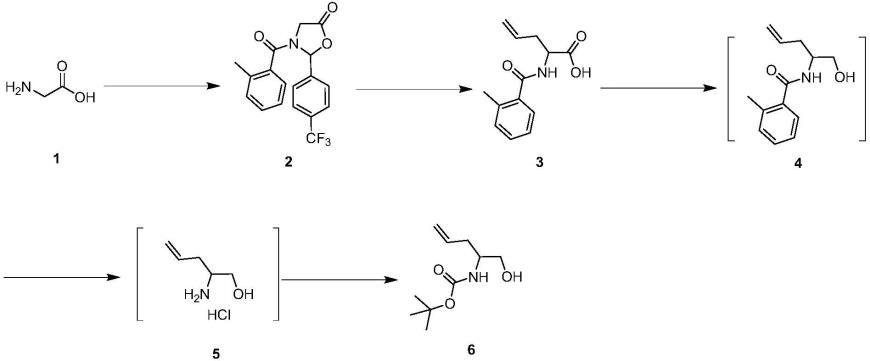
本发明提供了(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的合成方法,包括如下步骤:
[0012][0013]
(1)关环保护反应:化合物1与4-三氟甲基苯甲醛在溶剂中反应,之后再与2-甲基苯甲酰氯在碱的存在下反应得到化合物2;
[0014]
(2)烷基化反应:在溶剂中,化合物2在lda的存在下与烯丙基溴发生烷基化反应,之后在无机碱的存在下发生水解反应得化合物3;
[0015]
(3)工艺叠缩:在溶剂中,化合物3在n-甲基吗啡啉的存在下与对甲苯磺酰氯形成混酐,后经硼氢化钠还原得到化合物4;化合物4在酸的存在下发生水解反应,过滤得到化合物5;化合物5与二碳酸二叔丁酯在碱的存在下反应得到(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯。
[0016]
进一步地,步骤(1)中,所述溶剂为甲苯;所述碱为碳酸钾、碳酸钠、氢氧化钠或氢氧化钾。
[0017]
进一步地,步骤(2)中,所述溶剂为四氢呋喃;所述无机碱为氢氧化钠或氢氧化钾。
[0018]
进一步地,步骤(3)中,所述溶剂为四氢呋喃、1,4-二氧六环或乙二醇二甲醚;
[0019]
和/或,步骤(3)中,所述酸为盐酸或硫酸;
[0020]
和/或,步骤(3)中,所述碱为氢氧化钠、氢氧化钾、碳酸钠或碳酸钾。
[0021]
进一步地,
[0022]
步骤(1)中,化合物1、4-三氟甲基苯甲醛、2-甲基苯甲酰氯和碱的摩尔比为1.0:1.0~1.1:1.0~1.2:0.5~0.7;
[0023]
和/或,步骤(1)中,化合物1与溶剂的质量体积比为1.0:6.0~15.0。
[0024]
进一步地,
[0025]
步骤(2)中,化合物2、lda、烯丙基溴、无机碱的摩尔比为1.0:1.0~1.2:1.0~1.1:1.1~1.5;
[0026]
和/或,步骤(2)中,化合物2与溶剂的质量体积比为1.0:6.0~15.0。
[0027]
进一步地,
[0028]
步骤(3)中,化合物3、n-甲基吗啡啉、对甲苯磺酰氯与硼氢化钠的摩尔比为1.0:1.1~1.3:1.0~1.2:1.3~1.8;
[0029]
和/或,步骤(3)中,化合物3与溶剂的质量体积比为1.0:6.0~15.0。
[0030]
进一步地,
[0031]
步骤(3)中,化合物3与酸的质量体积比为1.0:5.0~10.0。
[0032]
进一步地,
[0033]
步骤(3)中,化合物3与二碳酸二叔丁酯的摩尔比为1.0:1.0~1.2;
[0034]
和/或,步骤(3)中,所述碱调节ph至8-9。
[0035]
进一步地,
[0036]
步骤(1)中,所述化合物1与4-三氟甲基苯甲醛反应温度为100~110℃回流;
[0037]
和/或,步骤(1)中,所述与2-甲基苯甲酰氯在碱的存在下反应时反应温度是0~10℃;
[0038]
和/或,步骤(2)中,所述烷基化反应的反应温度是-80~-70℃;
[0039]
和/或,步骤(2)中,所述水解反应的反应温度是0~10℃;
[0040]
和/或,步骤(3)中,所述与对甲苯磺酰氯形成混酐时反应温度是0~10℃;
[0041]
和/或,步骤(3)中,所述经硼氢化钠还原的反应温度是-10~0℃;
[0042]
和/或,步骤(3)中,所述水解反应的反应温度是95~105℃回流;
[0043]
和/或,步骤(3)中,所述化合物5与二碳酸二叔丁酯在碱的存在下反应的反应温度是10~30℃。
[0044]
本发明中,工艺叠缩是指直接将未经纯化后处理的粗产物投入下步反应,经过数步免除纯化过程(反应路线中可用符号[]表示)后再经分离纯化获取高纯度产物。工艺叠缩中的数步反应作为一个系统总体考虑。
[0045]
与现有技术相比,本发明的有益效果为:
[0046]
本发明提供了一种新的合成(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的方法,本发明合成方法所使用的原料价格低廉、易得,没有使用高危反应,所使用的设备要求简单,工艺叠缩,保护基可以回收,产物收率高、纯度高。可见本发明合成方法成本低,工艺简便,安全性好,同时放大生产效果好,适合大规模工业生产,解决了现有合成(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的方法不适合工业化大批量生产的问题,具有良好的应用前景。
[0047]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0048]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0049]
图1为实施例2放大生产制备得到的(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的hnmr图谱。
[0050]
图2为实施例2放大生产制备得到的(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的纯度检测结果。
具体实施方式
[0051]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0052]
本发明采用的合成方法的反应路线,如下所示:
[0053][0054]
实施例1、(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的合成方法
[0055]
1、化合物2的制备
[0056][0057]
将化合物1(100.0g,1.332mol,1.0eq)和4-三氟甲基苯甲醛(231.9g,1.332mol,1.0eq)加入甲苯(1000ml)中。氮气保护下,升温至100-110℃回流反应16h,通过分水器把水分出。将体系降温至0-10℃,准备两个滴加漏斗,同时滴加20%碳酸钾水溶液(551.3g,0.799mol,0.6eq)和2-甲基苯甲酰氯(226.5g,1.465mol,1.1eq),控温0-10℃,控体系ph=
6-9。滴毕,搅拌4h。过滤,滤饼干燥得到化合物2(386.2g,1.106mol),收率83%。
[0058]
2、化合物3的制备
[0059][0060]
将2m lda(157.5ml,0.315mol,1.1eq)和四氢呋喃(200ml)混合液在氮气保护下降温至-80~-70℃,滴加化合物2(100g,0.286mol,1.0eq)的四氢呋喃(500ml)溶液,控温-80~-70℃。滴毕,搅拌2h。将烯丙基溴(36.3g,0.300mol,1.05eq)溶于四氢呋喃(100ml)中,滴入,控温-80~-70℃。滴毕,搅拌3h。将反应液淬灭至水(800ml)中,加入氢氧化钠(13.7g,0.343mol,1.2eq),控温0-10℃搅拌3h。反应液用正庚烷(600ml x2)萃取除杂。水相用浓盐酸调节ph=2,用乙酸乙酯(600ml x3)萃取。有机相合并,用饱和食盐水(600ml)洗涤,有机相浓缩得到化合物3(49.4g,0.212mol),收率74%。
[0061]
3、化合物6的制备
[0062][0063]
向化合物3(50g,0.214mol,1.0eq)的四氢呋喃(250ml)溶液中,加入n-甲基吗啡啉(26.0g,0.257mol,1.2eq),氮气保护下降至0~10℃。将对甲苯磺酰氯(44.8g,0.235mol,1.1eq)溶于四氢呋喃(150ml)中滴入,控温0~10℃。滴毕,搅拌2h,之后过滤(固体舍弃),将滤液降温至-10~-5℃。将硼氢化钠(12.2g,0.321mol,1.5eq)溶于0.4%氢氧化钠水溶液(100ml)中,滴入反应液中,控温-10~0℃。滴毕,缓慢升至室温,搅拌1h。将反应液缓慢淬灭至浓盐酸(50ml)中,减压浓缩掉四氢呋喃。
[0064]
加入浓盐酸(250ml),升至95~105℃回流反应12h,降至0~10℃,过滤。
[0065]
滤饼加入水(400ml)中,将二碳酸二叔丁酯(51.2g,0.235mol,1.1eq)溶于甲醇(100ml)中滴入,用20%氢氧化钠调节ph=8-9,控温10~30℃。滴毕,继续反应8h。反应液减压浓缩掉甲醇,用二氯甲烷(300ml x3)萃取,有机相合并用水(300ml)洗涤,10%柠檬酸(300ml)洗涤,再用水(300ml)洗涤。有机相浓缩得到化合物6(30.6g,0.152mol),收率71%,纯度为99.9%(elsd检测)。化合物6即为(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯。
[0066]
实施例2、(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的工业化生产
[0067]
1、化合物2的放大生产
[0068][0069]
在1500l反应釜中加入甲苯(600l),将化合物1(50.0kg,666.0mol,1.0eq)和4-三氟甲基苯甲醛(116.0kg,666.0mol,1.0eq)一次加入。氮气保护下,升温至100-110℃回流反应16h,通过分水器把水分出。将体系降温至0-10℃,准备两个蠕动泵,同时滴加20%碳酸钾水溶液(275.6kg,399.6mol,0.6eq)和2-甲基苯甲酰氯(113.3kg,732.6mol,1.1eq),控温0-10℃,控体系ph=6-9。滴毕,搅拌8h。离心,滤饼干燥得到化合物2(200.1kg,572.7mol),收率86%。
[0070]
2、化合物3的放大生产
[0071][0072]
1000l低温釜氮气置换三次,抽入四氢呋喃(100l)和2m lda(157.5l,315.0mol,1.1eq)。氮气保护降温至-80~-70℃,滴加化合物2(100kg,286.0mol,1.0eq)的四氢呋喃(600l)溶液,控温-80~-70℃。滴毕,搅拌2h。将烯丙基溴(36.3kg,300mol,1.05eq)滴入,控温-80~-70℃。滴毕,搅拌3h。将反应液淬灭至水(800l)中,加入氢氧化钠(13.7kg,343mol,1.2eq),控温0-10℃搅拌6h。反应液用正庚烷(600l x2)萃取除杂。水相用浓盐酸调节ph=2,用乙酸乙酯(600l x3)萃取。有机相合并,用饱和食盐水(600l)洗涤,有机相浓缩得到化合物3(53.4kg,228.8mol),收率80%。
[0073]
3、化合物6的放大生产
[0074][0075]
向1500l反应釜中加入四氢呋喃(250l)和化合物3(50kg,214.0mol,1.0eq),加入n-甲基吗啡啉(26.0kg,257.2mol,1.2eq),氮气保护下降至0~10℃。将对甲苯磺酰氯(44.8kg,235.1mol,1.1eq)溶于四氢呋喃(150l)中滴入,控温0~10℃。滴毕,搅拌2h,之后过滤(固体舍弃),将滤液降温至-10~-5℃。将硼氢化钠(12.2kg,321.5mol,1.5eq)溶于0.4%氢氧化钠水溶液(100l)中,滴入反应液中,控温-10~0℃。滴毕,缓慢升至室温,搅拌1h。将反应液缓慢淬灭至浓盐酸(50l)中,减压浓缩掉四氢呋喃。
[0076]
加入浓盐酸(300l),升至95~105℃回流反应12h,降至0~10℃,过滤。
[0077]
滤饼加入水(400l)中,将二碳酸二叔丁酯(51.2kg,235.1mol,1.1eq)溶于甲醇
(100l)中滴入,用20%氢氧化钠调节ph=8-9,控温10~30℃。滴毕,继续反应8h。反应液减压浓缩掉甲醇,用二氯甲烷(300l x3)萃取,有机相合并用水(300l)洗涤,10%柠檬酸(300l)洗涤,再用水(300l)洗涤。有机相浓缩得到化合物6(33.6kg,166.9mol),收率78%,纯度为99.9%(elsd检测)。化合物6即为(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯。化合物6的hnmr图谱如图1所示:hnmr(400mhz,cdcl3):5.72-5.83(m,1h),5.08-5.13(m,2h),4.74(br,1h),3.57-3.67(m,3h),2.76(br,1h),2.21-2.32(m,2h),1.43(s,9h).。化合物6的纯度检测结果如图2所示。
[0078]
实施例2可知,本发明合成(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的方法工业化放大生产后依然可以取得优异的收率和纯度。
[0079]
综上所述,本发明提供了一种新的合成(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的方法,本发明合成方法所使用的原料价格低廉、易得,没有使用高危反应,所使用的设备要求简单,工艺叠缩,保护基可以回收,产物收率高、纯度高。可见本发明合成方法成本低,工艺简便,安全性好,同时放大生产效果好,适合大规模工业生产,解决了现有合成(1-羟基戊-4-烯-2-基)氨基甲酸叔丁酯的方法不适合工业化大批量生产的问题,具有良好的应用前景。
[0080]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。