抗病毒前体药物及其药物组合物
1.相关申请的交叉引用
2.本技术要求于2019年9月11日提交的美国临时申请序列号62/898,679的权益,其全部内容通过引用并入本文。
技术领域
3.本发明涉及可用于治疗获得性免疫缺陷综合征(aids)的抗病毒化合物和组合物。
背景技术:
4.由式i表示的4'-乙炔基-2-氟-2'-脱氧腺苷(efda)(mk-8591):
[0005][0006]
是一种核苷类似物,可作为核苷逆转录酶的有效抑制剂(current opinion in hiv and aids 2018,13,294-299),可用作治疗和暴露前预防hiv-1感染的抗逆转录病毒药物。efda在细胞中代谢为活性三磷酸合成代谢物(efda-tp),该代谢物抑制hiv逆转录酶。
[0007]
然而,efda具有相对高的水溶性和相对短时间过程的血药浓度。结果是当将efda施用于病人用于治疗人类免疫缺陷病毒(hiv)感染或用于暴露前预防时,efda只能提供有限的病毒抑制持续时间。因此,需要能够延长给药后病毒抑制持续时间的配方。本发明的化合物和药物制剂满足了这种需要。
技术实现要素:
[0008]
本发明的抗病毒化合物是efda的二酯并且在水中具有有限的溶解度。尽管efda在生理ph下具有0.877mg/ml的水溶性,但本发明的efda二酯在生理ph下具有小于0.03mg/ml的水溶性,优选地小于0.002mg/ml。这些efda的二酯是结晶的,可用于提供延长的hiv抑制持续时间,并且可以在药学上可接受的载体中作为悬浮液不经胃肠道施用。人类受试者的优选预防剂量是约80mg至约800mg范围内的efda二酯,以约6个月的间隔,以每剂约0.5ml至约4ml的剂量体积不经胃肠道施用。人类患者的优选治疗剂量是约80mg至约800mg范围内的efda二酯,以约3个月的间隔,以每剂约0.5ml至约4ml的剂量体积不经胃肠道施用。
[0009]
本发明包含抗病毒化合物的不经胃肠道制剂可以是包含抗病毒化合物以及药学上可接受辅料的干燥制剂或抗病毒化合物在水性或油基介质中的稳定悬浮液。
附图说明
[0010]
附图中,
[0011]
图1示出了efda的x射线粉末衍射图(xprd)。
[0012]
图2示出了由差示扫描量热法(dsc)和热重分析(tga)提供的efda数据。
[0013]
图3示出了化合物2的dsc和tga数据。
[0014]
图4示出了化合物3的xprd数据。
[0015]
图5示出了化合物3的dsc数据。
[0016]
图6示出了化合物3的tga数据。
[0017]
图7示出了化合物5的xprd数据。
[0018]
图8示出了化合物5的dsc数据。
[0019]
图9示出了化合物5的tga数据。
[0020]
图10以图形形式示出了表2中提供的数据。
[0021]
图11以图表形式示出了表3中提供的数据。
[0022]
图12以图形形式示出了表4中的数据。
[0023]
图13以图形形式示出了实施例10的数据。
[0024]
图14示出了化合物2的xprd数据。
[0025]
图15示出了化合物6的xprd数据。
具体实施方式
[0026]
由式(ii)表示的二酯实现了延长的体内病毒抑制:
[0027][0028]
其中r1和r2独立地为-c(=o)r3,且r3是由异丙基、3-戊基、环戊基和苯甲基组成的组中的一员。上述二酯是通过使efda与所需的酸或酸酐反应并将二酯作为结晶化合物回收来制备的。以下实施例说明了优选二酯的制备。
[0029]
(2r,3s,5r)-5-(6-氨基-2-氟-9h-嘌呤-9-基)-2-乙炔基-2-((异丁酰氧基)甲基)四氢呋喃-3-基异丁酸酯的制备
[0030][0031]
在室温下往无水二甲基甲酰胺(dmf)(100ml)中efda(化合物1)(3g,6.8mmol,1当量(equiv.))、4-二甲基氨基吡啶(dmap)(499mg,2.73mmol,0.4当量(equiv.))的混合物中滴加异丁酸(8.4g,27.3mmol,6当量(equiv.))。在室温下搅拌反应5h。由于在延长反应时间时观察到nh2基团偶尔发生烷基化,因此通过lc-ms监测反应。然后过滤反应混合物以除去副产物尿素。使用乙腈冲洗反应混合物。此后,将反应混合物用水洗涤两次并用盐水洗涤一次,然后将溶剂干燥、过滤并减压蒸发。使用含60-70%乙酸乙酯(etoac)的己烷通过硅胶柱层析法纯化所得粗物质,得到呈玻璃状固体的化合物2。将获得的玻璃状固体分散在最少量的异丙醇中,然后将其旋转蒸发以获得纯的化合物2,为白色固体(2.5g,85%得率)。lc-ms(esi ):m/z 434.49[m h]
。
[0032]
将约250mg/g的化合物2悬浮在水性的0.25%cmc-na/0.5%吐温-80中(26g可注射性)中,并在40℃/75%相对湿度下测试2周的稳定性。图14提供了来自孵育悬浮液的化合物6的xprd数据,孵育前(下图)、一周后(中图)和两周后(上图),表明化合物在悬浮液中保持良好的结晶度。
[0033]
(2r,3s,5r)-5-(6-氨基-2-氟-9h-嘌呤-9-基)-2-(((2-乙基丁酰基)氧基)甲基)-2-乙炔基四氢呋喃-3-基2-丁酸乙酯的制备
[0034][0035]
将无水乙腈(mecn)(43ml)中efda(1g,3.4mmol,1equiv.)、2-乙基丁酸酐(4.4g,20.4mmol,6equiv.)、三乙醇胺(tea)(3.8ml,27.2mmol,8equiv)的混合物冷却至0℃,并在0℃下加入4-二甲基氨基吡啶(dmap)(83mg,0.68mmol,0.2equiv)。将所得混合物在0℃搅拌0.5h,然后在室温下搅拌5h。由于在延长反应时间时观察到nh2基团偶尔发生烷基化,因此通过lc-ms监测反应。用甲醇淬灭反应混合物,并减压蒸发溶剂。使用含60-70%乙酸乙酯
(etoac)的己烷通过硅胶柱层析法纯化所得粗物质,得到呈玻璃状固体的化合物3。将获得的玻璃状固体分散在最少量的异丙醇中,然后将其旋转蒸发以获得纯的化合物3,为白色固体(1.33g,80%得率)。lc-ms(esi ):m/z 490.56[m h]
。
[0036]
(2r,3s,5r)-5-(6-氨基-2-氟-9h-嘌呤-9-基)-2-(((环戊烷羰基)氧基)甲基)-2-乙炔基四氢呋喃-3-基环戊烷羧酸酯的制备
[0037][0038]
除了使用环戊酸代替异丁酸以外,化合物4通过使用化合物2所遵循的程序来制备。lc-ms(esi ):m/z 486.44[m h]
。
[0039]
(2r,3s,5r)-5-(6-氨基-2-氟-9h-嘌呤-9-基)-2-乙炔基-2-((2-苯基乙酰氧基)甲基)四氢呋喃-3-基2-苯乙酸酯的制备
[0040][0041]
将无水乙腈(mecn)(22ml)中efda(499mg,1.7mmol,1equiv.)、2-苯乙酸酐(2.6g,10.2mmol,6equiv.)、三乙醇胺(tea)(1.9ml,13.6mmol,8equiv.)的混合物冷却至0℃,并在0℃下加入4-二甲基氨基吡啶(dmap)(42mg,0.34mmol,0.2equiv)。在0℃下搅拌反应0.5h,然后在室温搅拌3h。由于在延长反应时间时观察到nh2基团偶尔发生烷基化,因此通过lc-ms监测反应。用甲醇淬灭反应混合物,减压蒸发溶剂。使用含60-70%乙酸乙酯(etoac)的己烷通过硅胶柱层析法纯化所得粗物质,得到呈玻璃状固体的化合物5。将获得的玻璃状固体分散在最少量的异丙醇中,然后将其旋转蒸发以获得纯的化合物5,为白色固体(694mg,77%得率)。lc-ms(esi ):m/z 530.52[m h]
。
[0042]
表1显示了通过核磁共振(nmr)和液相色谱-质谱(lc-ms)获得的化合物表征数据。(2r,3s,5r)-5-(6-氨基-2-氟-9h-嘌呤-9-基)-2-((苯甲酰氧基)甲基)-2-乙炔基四氢呋喃-3-基苯甲酸酯,化合物6
[0043]
化合物6通过使用类似于制备化合物2的程序和使用4.5equiv的相应酸来制备。
lc-ms(esi ):m/z 502.41[m h]
。将约275mg/g的化合物6悬浮在水性的0.25%cmc-na/0.5%吐温-80中(26g可注射性),并在40℃/75%相对湿度下测试稳定性。图15提供了来自孵育悬浮液的化合物6的xprd数据,孵育前(下图)、一周后(中图)和两周后(上图),表明化合物在悬浮液中保持良好的结晶度。
[0044]
表1:优选的化合物
[0045]
[0046][0047]
前述讨论和实施例都是说明性的,不应被视为限制性的。在本发明精神范围内的其他变体是可能的并且将容易地呈现给本领域技术人员。
[0048]
稳定晶型的合成
[0049]
实施例1
[0050]
在搅拌下将化合物2(~200mg)完全溶解在最少量的丙酮中。随后在室温下缓慢蒸
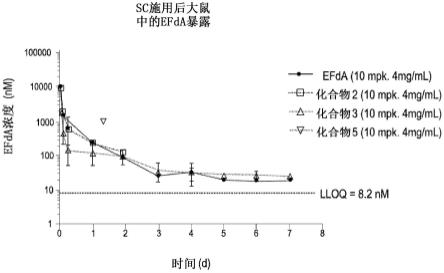
发溶剂。得到呈白色粉末的重结晶样品。也可以使用其他溶剂,包括乙酸乙酯、甲醇和四氢呋喃。
[0051]
本发明化合物制剂的总体实施例
[0052]
所有制剂方案均产生具有26g可注射性的稳定水性悬浮液。
[0053]
efda制剂
[0054]
efda被研磨并通过#80筛子过筛。将预制的0.25%羧甲基纤维素钠(sodium cmc)与0.1%聚氧乙烯(20)山梨醇酐单油酸酯(吐温-80)溶液添加到约300mg的efda(化合物1)中,以提供约1g(300mg化合物1 约700mg聚合物溶液)的最终悬浮液制剂(约300mg/g)。然后将悬浮液在冰浴中超声处理10min。该制剂的密度为1.064g/ml并提供319.2mg/ml的efda浓度。
[0055]
实施例2
[0056]
将重结晶的化合物2研磨并通过用于金属丝编织筛布的美国标准筛系列80号筛(标称筛孔0.180mm)过筛。将250mg的化合物2放入合适的容器中,加入预制的0.25%羧甲基纤维素钠(cmc)与0.1%吐温-80溶液,以得到1g(250mg化合物2 约750mg聚合物溶液)的最终制剂(~250mg/g)。然后将悬浮液在冰浴中探针式超声处理5min(超声处理时间:5min;脉冲幅度:20;脉冲开启时间:30s;脉冲关闭时间:20s)。
[0057]
实施例3
[0058]
将化合物3研磨并通过用于金属丝编织筛布的美国标准筛系列80号筛(标称筛孔0.180mm)过筛。将250mg的化合物3放入合适的容器中,并加入预制的0.25%cmc钠与0.5%吐温-80溶液,以得到1g(250mg化合物3 约750mg聚合物溶液)的最终悬浮液(~250mg/g)。然后将悬浮液在冰浴中探针式超声处理5min(超声处理时间:5min;脉冲幅度:20;脉冲开启时间:30s;脉冲关闭时间:20s)。悬浮液的密度为1.004g/ml。该悬浮液含有250.88mg/ml的化合物3(~150mg/ml efda)。
[0059]
实施例4
[0060]
将化合物5研磨并通过用于金属丝编织筛布的美国标准筛系列80号筛(标称筛孔0.180mm)过筛。将200mg的化合物5置入合适的容器中,并加入预制的0.25%cmc钠与0.5%吐温-80的溶液,以得到1g(200mg化合物5 约800mg聚合物溶液)的最终悬浮液(~200mg/g)。然后将悬浮液在冰浴中探针式超声处理5min((超声处理时间:5min;脉冲幅度:20;脉冲开启时间:30s;脉冲关闭时间:20s)。该悬浮液的密度为1.047g/ml。该悬浮液含有209.4mg/ml的化合物5(~115.97mg/ml efda)。
[0061]
药代动力学(pk)研究
[0062]
动物:动物(雄性sd大鼠~200-250g和雄性恒河猴~2-3kg)获自经批准的供应商(斯莱克实验动物有限责任公司,中国上海和/或天勤生物科技有限公司,中国湖北省武汉市)。
[0063]
适应/隔离:到达后,由兽医人员或其他授权人员评估动物的一般健康状况。在进行研究之前,使动物适应至少3天。
[0064]
动物饲养:在适应期间将动物分组饲养,在研究期间单独饲养。控制动物房环境(目标条件:温度18至26℃,相对湿度30至70%,12h人工光照和12h人工黑暗)。每天监测温度和相对湿度。
[0065]
动物插管:否
[0066]
动物在施用药物前禁食至少12h。给药后4h,所有动物都可以自由获取经认证的啮齿动物和非啮齿动物饮食(货号#m01-f,斯莱克实验动物有限责任公司,中国上海)。
[0067]
水在提供给动物自由饮用之前进行高压灭菌。对水进行定期分析并将结果存档。没有在饮食或水中检测到已知的预计会干扰研究目的、实施或结果的污染物。
[0068]
1.剂量制剂
[0069]
sc制剂:在给药当天根据以上实施例2-5和表2-4中描述的程序制备悬浮液。在悬浮液制备的4h内施用于动物。将每种制备的悬浮液的两个20μl等分试样转移到1.5ml的聚丙烯微量离心管中,并通过lc/uv或lc-ms/ms进行剂量验证。
[0070]
2.剂量施用
[0071]
按照设备标准操作程序(sop)通过皮下注射(sc)施用悬浮液。
[0072]
3.样品采集
[0073]
在每个时间点从大鼠隐静脉采集约200μl血液,从恒河猴采集约0.5ml血液。将所有血样转移到含有4μl k2edta(0.5m)作为抗凝剂的微量离心管中,并置于湿冰上直至对血浆进行处理。
[0074]
4.血液/血浆处理
[0075]
血液:在采集后半小时内,通过在约4℃、3000g下离心15min来处理血液样本以获得血浆。血浆样品储存在聚丙烯管中,在干冰上快速冷冻并保持在-70
±
10℃直至进行lc/ms/ms分析。
[0076]
5.样本分析
[0077]
剂量浓度验证
[0078]
■
从每种制备的悬浮液的中间位置收集等分悬浮液,一式两份。
[0079]
■
每个等分悬浮液中的活性成分浓度通过lc/uv或lc/ms/ms方法测定
[0080]
生物分析方法和样品分析
[0081]
■
用于定量测定相应生物基质中活性成分(所测试的化合物)的lc-ms/ms方法是在非glp合规性下开发的。
[0082]
■
一条包含8个非零校准标准品的校准曲线适用于该包括定量下限(lloq)的方法。
[0083]
■
该方法应用了一组由低、中、高浓度组成的质量控制(qc)样品。
[0084]
■
研究样品分析将使用lc-ms/ms方法与一组校准标准品和两组qc样品同时进行(如果样品数量超过48个,则应用带有2组qc样品的两条校准曲线)。
[0085]
■
验收标准:
[0086]
线性:至少6个校准标准品被回算到血浆中其标称值的
±
20%以内
[0087]
准确度:6个qc样品中至少有4个被回算到血浆中其标称值的
±
20%以内。
[0088]
特异性:单个空白基质中的平均计算浓度应为lloq的0.5倍。
[0089]
灵敏度:定量下限(lloq)的目标是1~3ng/ml。
[0090]
残留物:在最高标准品上样后接下来的单个空白基质样品中计算的平均残留物浓度应为lloq。如果残留物不符合标准,则应按照内部生物分析sop估计残留物的百分比。
[0091]
6.数据分析
[0092]
使用phoenix winnonlin 6.3软件程序通过非房室方法分析血浆浓度对时间的数据。生成了c
max
、t
max
、t
1/2
、auc
(0-t)
、auc
(0-inf)
、mrt
(0-t)
、mrt
(0-inf)
、%f和血浆浓度-时间曲线图。
[0093]
实施例5:
[0094]
包括作为对照的efda(1)在内的几种前体药物通过皮下注射途径施用于大鼠进行单剂量pk研究。以在0.5%cmc-na与0.5%吐温-80中的水性悬浮液的形式,所有动物都注射了10mg/kg当量剂量和4mg/ml浓度的efda。虽然观察到类似的暴露,但所有化合物2、3和5显示的血浆efda水平高于定量下限(lloq)超过一周,c
max
远低于efda。
[0095]
表2示出了以10mg/kg当量剂量的efda皮下(sc)施用化合物1、2、3和5后大鼠的pk数据。数据以图形形式显示在图10中。
[0096]
表2:sc大鼠的pk:efda、化合物1、2、3和5的efda水平
[0097][0098][0099]
实施例6:
[0100]
优化后,使用100mg/kg的高当量剂量分别以各化合物的当量浓度再次进行sc大鼠的pk研究,化合物3以120mg/ml efda、化合物5以116mg/ml efda,efda以319mg/ml efda。化合物3提供的c
max
比efda延迟且低100倍。还观察到延长的半衰期和平均停留寿命,使得化合物3和化合物5适合用于预防。
[0101]
表3a和表3b显示了sc施用化合物1、3和5后的大鼠pk数据。化合物3的数据以图形形式显示在图11中。
[0102]
表3a:sc大鼠的pk:efda和化合物3的efda水平
[0103][0104]
表3b:sc大鼠的pk:efda和化合物5的efda水平
[0105][0106]
实施例7:
[0107]
经大鼠的pk分析后,重点转移到非啮齿动物,即恒河猴。通过皮下注射途径施用当量剂量为50mg/kg efda的化合物5和efda,以进行单剂量恒河猴pk研究。水性悬浮液包含0.25%cmc-na与0.1%/0.5%吐温-80,化合物5和efda的当量浓度分别为116mg/ml和319mg/ml的efda。观察到化合物5的血浆efda水平高于lloq超过一个月,c
max
比efda本身低24倍。
[0108]
表4显示了化合物1和5在以50mg/kg当量剂量的efda皮下(sc)施用后恒河猴的pk数据。数据以图形形式显示在图12中。
[0109]
表4:sc恒河猴的pk:在血浆中施用efda或化合物5后的efda血浆水平
[0110][0111][0112]
本发明化合物的悬浮介质可以是水性载体,例如注射用水(wfi)或植物油载体,例如芝麻油、橄榄油等。悬浮介质还可以含有药学上可接受的辅料,例如非离子表面活性剂、悬浮剂或絮凝剂、防腐剂、缓冲剂、毒性调节剂、螯合剂、抗氧化剂等。
[0113]
对于预防,人类受试者的优选预防剂量在约80mg至约800mg的范围内,以每剂约0.5ml至约4ml的剂量体积以约6个月(半年)的间隔不经胃肠道施用抗病毒化合物。
[0114]
对于人类患者的治疗,本发明的抗病毒化合物的有效量优选为约80mg至约800mg,剂量体积为每剂约0.5ml至约4ml,更优选地有效量为约200mg至约400mg,给药方案为期3个月。然而,根据具体给药方案中施用剂量之间的时间间隔,给药方案可以有所不同。
[0115]
如本说明书和在权利要求中使用的术语“有效量”是指足以抑制hiv逆转录酶、抑制hiv复制、在施用后发挥预防作用和/或发挥治疗作用的抗病毒化合物的量。
[0116]
关于所要保护的治疗方法的术语“施用”及其变体,例如“施用化合物”是指向患者提供抗病毒化合物并且包括自我施用以及由另一个人向患者施用该化合物。
[0117]
优选地,适用于注射的不经胃肠道悬浮剂包含基于悬浮剂重量的约3-45重量%的本发明抗病毒化合物。优选的粒度不大于约50微米(μm),更优选地平均粒度在约6μm至约15μm的范围内。
[0118]
优选的絮凝剂或悬浮剂是线型聚合物,特别是取代纤维素,例如甲基纤维素、羧甲基纤维素(cmc)、羟丙基纤维素、羟丙基甲基纤维素等。
[0119]
优选的表面活性剂是非离子表面活性剂。特别优选的表面活性剂是聚氧乙烯(20)山梨醇酐单油酸酯(吐温-80)。
[0120]
除了不经胃肠道的剂型之外,本发明化合物也可以以口服剂型和作为植入物施用。
[0121]
含有本发明化合物的剂型还可以包括额外的抗hiv和/或抗hbv药剂,例如卡博特
韦(cabotegravir)、杜鲁特韦(dolutegravir)、多拉维林(doravirine)、埃替格韦(elvitegravi)、勒西韦林(lersiverine)、富马酸泰诺福韦酯(tenofovir disoproxil fumarate)、富马酸替诺福韦艾拉酚胺(tenofovir alafenamide fumarate)和拉米夫定(lamivudine)等。
[0122]
在各种实施方案中,本发明进一步提供了在患者中预防病毒血症或治疗病毒感染的方法,其中逆转录酶的抑制在医学上是有指征的,包括向患者施用有效量或浓度的式(ii)化合物。更具体地,式(ii)化合物可以以提供efda从这些前体药物中缓慢或受控或持续释放的制剂形式施用。更具体地,式(ii)化合物可以配制成水性悬浮液、溶液,并且可以包封在用于缓释的颗粒中,包括plga和本领域已知的其他此类材料。更具体地,病毒感染可以由hiv或hbv引起。这些前体药物的给药途径可以包括但不限于口服、不经胃肠道和从植入物(药物递送组合物和装置)中释放。在用于治疗或预防病毒感染的方法中,该方法还可以包括额外的抗hiv和/或抗hbv药剂,包括但不限于卡博特韦、杜鲁特韦、多拉维林、埃替格韦、勒西韦林、富马酸泰诺福韦酯、富马酸替诺福韦艾拉酚胺和拉米夫定等。
[0123]
实施例8
[0124]
在含有400mg/gm微粉化的化合物5的组成物中,用0.3%和0.5%的甲基纤维素优化表面活性剂的浓度。用不同浓度的吐温-80(0.1、0.2和0.3%)制备制剂。在储存10天后,未观察到表面活性剂浓度对制剂的粘度、流动和重悬时间有显着影响。观察结果如表5所示。
[0125]
表5:用400mg/gm的化合物5对表面活性剂浓度优化
[0126][0127]
粘度: 轻微粘性, 粘性, 非常粘性, 半固体
[0128]
流量: 无流动, 流动较少, 流动良好, 流动非常好√:可注射
[0129]
实施例9
[0130]
用0.3%甲基纤维素和0.2%吐温-80浓度制备一批(40gm)的化合物5悬浮液。尽管聚合物和表面活性剂的浓度用40重量%的药物浓度进行了优化,但在加速和长期稳定性研究期间,还制备了具有35重量%药物浓度的批次。表6中给出了在稳定室中装载的制剂的组成。下面给出了制剂制备和组成的详细制造程序。
[0131]
表6:制剂组成
[0132][0133][0134]
将甲基纤维素缓慢加入到玻璃瓶中所需量的注射用水(wfi)中,同时持续搅拌。使用磁力搅拌器搅拌所得混合物直至溶液澄清且没有任何结块。将吐温-80(0.08g)加入到获得的溶液中并充分搅拌。
[0135]
使用磁力搅拌器在1400rpm的剧烈搅拌下将微粉化的化合物5(14g;平均粒度11μm)缓慢加入到制备的聚合物、表面活性剂溶液中。在完全添加化合物5后,将获得的悬浮液搅拌(600rpm)20分钟以均匀分散颗粒。
[0136]
对制备的制剂进行各种理化性质的表征,例如化验、ph、%纯度、再分散性、可注射性、多态形式和粒度。结果如表7所示。
[0137]
表7:制备制剂的理化表征
[0138]
测试参数 外观白色分散体ph5.78hplc化验(%标示量)108.8%%纯度99.34%再分散性有可注射性有多态形式与api相同平均粒度14μm
[0139]
根据表7所示的特性,表6所示的制剂被认为是可接受的。
[0140]
实施例10
[0141]
将efda和化合物3和5的水性悬浮液皮下施用于恒河猴,定期采集外周血单核细胞(pbmc),并评估药代动力学数据。两种化合物的观察结果如表8、9和10所示,化合物5的观察结果如图13所示。
[0142]
表8:sc恒河猴的pk:施用化合物5或efda后efda-tp的pbmc水平
[0143]
[0144][0145]
表9:sc恒河猴的pk:施用化合物3后efda血浆水平和efda-tp的pbmc水平
[0146][0147]
表10:sc恒河猴的pk:血浆中化合物3和5的efda水平
[0148]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。