1.本发明涉及一种瑞卢戈利中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯)的制备方法。
背景技术:
[0002]
瑞卢戈利是一种口服种口服促性腺激素释放激素受体拮抗剂,可以阻断促性腺激素释放激素受体并减少睾丸睾酮的生成,睾丸睾酮是一种已知的刺激前列腺癌生长的激素。2020年先在日本上市,获批适应症为子宫肌瘤,剂量为40mg,2021年在美国上市,获批适应症为前列腺癌,剂量为120mg。其原料药在合成过程中使用到了一种关键中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯)(以下称化合物b1)
[0003]
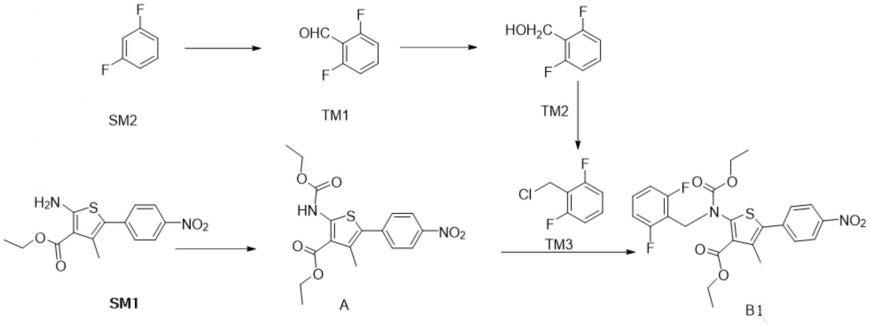
专利cn109053766报道了合成化合物b1的方法,路线如下:
[0004][0005]
该路线的不足之处是:合成路线长,尤其是tm3制备过程中,后处理操作复杂,产生的三废较多,对于降低成本不利。
[0006]
由于该品种适应症不断的扩大,而且剂量大,原料药和中间体的需求量大,非常有必要开发一条高效,成本低,污染小的工业化生产路线。
技术实现要素:
[0007]
本发明的目的在于提供一条高效,成本低,污染小,生产瑞卢戈利中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯)的制备方法。
[0008]
本发明的目的通过如下技术方案实现:
[0009]
一种瑞卢戈利中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯)的制备方法,它包括以下步骤:
[0010]
(1)2,6-二氟苯甲醛(tm1)的合成:本步骤的合成可按照文献tetrahedron letters 54(2013),6053-6056的方法完成,所述2,6-二氟苯甲醛(tm1)的结构式如下:
[0011][0012]
(2)化合物a1的合成:化合物sm1和化合物tm1发生醛胺缩合,生成希夫碱,希夫碱在还原试剂作用下生成化合物a1;其中,化合物sm1以及化合物a1的结构式如下:
[0013][0014]
(3)化合物b1的合成:将步骤(2)所得的化合物a1和氯甲酸乙酯反应得到化合物b1,即所述中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯);其中,化合物b1的结构式如下:
[0015][0016]
本发明瑞卢戈利中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯)合成的具体反应路线如下:
[0017][0018]
较之现有技术而言,本发明的优点在于:。
[0019]
1.本发明工艺路线步骤短,由原来的5步反应,缩短到3步反应。并且每一步反应收率高,经济效应得到很大提高。
[0020]
2.本发明工艺路线避免原工艺路线制备tm3过程中,大量的用蒸馏操作步骤,而导致的操作繁琐、纯化分离相对困难的问题。
[0021]
3.传统的合成路线中,要用到tm3的制备过程,而该过程需要用到大量的酸,而本发明的工艺方法未使用tm3制备过程,相比传统工艺大大降低了废酸溶液的处理,有效节约了污水处理成本,是一种环境友好型生产工艺。
附图说明
[0022]
图1是本发明实施例一中化合物tm1的核磁谱图。
[0023]
图2是本发明实施例一中化合物a1的核磁谱图。
具体实施方式
[0024]
下面结合说明书附图和实施例对本发明内容进行详细说明:
[0025]
一种瑞卢戈利中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯)的制备方法,它包括以下步骤:
[0026]
(1)2,6-二氟苯甲醛(tm1)的合成:本步骤的合成按照文献tetrahedron letters 54(2013),6053-6056的方法完成;其中,2,6-二氟苯甲醛的结构式如下:
[0027][0028]
(2)化合物a1的合成:化合物sm1和化合物tm1发生醛胺缩合,生成希夫碱,希夫碱在还原试剂作用下生成化合物a1;其中,化合物sm1以及化合物a1的结构式如下:
[0029][0030]
(3)化合物b1的合成:将步骤(2)所得的化合物a1和氯甲酸乙酯反应得到化合物b1,即所述中间体2-[(2,6-二氟苄基)乙氧基羰基氨基]-4-甲基-5-(4-硝基苯基)噻吩-3-甲酸乙酯);其中,化合物b1的结构式如下:
[0031][0032]
步骤(1)的具体操作方法为:在500ml的三口瓶中,加入间二氟甲苯,tmeda,dipa和thf。降低温度至-70~78℃,然后滴加丁基锂,滴加完毕后在此温度条件下反应1-6h(更优
选为2-3h),然后慢慢滴加dmf,在此温度条件下继续反应1-2h,将反应液升到室温,加入饱和的nh4cl溶液,再加入乙酸乙酯,萃取分液,水层用乙酸乙酯洗涤2遍,合并有机层,有机层浓缩得到tm1。
[0033]
其中,所述二氟甲苯、tmeda、dipa、丁基锂以及dmf的摩尔比为1:1.1~2.0:0.02~0.12:1.1~2.0:1.0~2.0.
[0034]
步骤(2)的具体操作方法为:在氮气保护下,在三口瓶中加入sm1、tm1和有机溶剂a、在温度10-100℃(更优选为60-70℃)条件下反应1-12h(更优选为5-6h),反应完毕后,将反应液冷却至5-10℃,在此温度条件下慢慢加入还原试剂,然后升温,在温度30-90℃(更优选为40-50℃)条件下反应2-10h(更优选为5-6h),反应完毕后,慢慢加入2n hcl调节ph值接近7,然后加入dcm,萃取分液,水层用dcm萃取2遍,合并有机层,有机层浓缩干燥得到固体a1;
[0035]
其中,化合物sm1、化合物tm1以及还原试剂的摩尔比为1:1.0~2.0:1.1~1.5。
[0036]
所述有机溶剂a为甲苯、甲醇或乙醇。
[0037]
所述还原试剂为硼氢化钠、硼氢化钾中的一种。
[0038]
步骤(3)的具体操作方法为:在氮气保护下,在三口瓶中加入化合物a1和有机溶剂b,在0-10℃条件下,慢慢滴加氯甲酸乙酯,滴加完毕后升温至50-130℃(更优选为105-115℃),在此温度下反应1-24h(更优选为3-4h),反应完毕后,降低温度至45-55℃,在此温度条件下滴加乙醇;然后自然降温到20-25℃,在此温度条件下搅拌1-2h,过滤,滤饼用乙醇洗涤,真空干燥得到固体化合物b1。
[0039]
所述有机溶剂b为甲苯、thf、二氧六环,更优选为甲苯。
[0040]
其中,反应时化合物a1、氯甲酸乙酯以及乙醇的摩尔比为1:1~2:1~2,更优选为1:1.1:1.1。
[0041]
实施例一:
[0042]
实施例1
[0043]
1.1 2,6-二氟苯甲醛的合成
[0044]
在500ml的三口瓶中,加入11.4g(0.1mol)间二氟甲苯,12.8g(0.11mol)tmeda,0.671g(0.005mol)dipa和thf。降低温度至-70~78℃,然后滴加7.0g(0.11mol)丁基锂,滴加完毕后在此温度条件下反应2-3h,然后慢慢滴加10.9g(0.15mol)dmf,在此温度条件下继续反应1-2h,将反应液升到室温,加入饱和的nh4cl溶液,再加入乙酸乙酯,萃取分液,水层用乙酸乙酯洗涤2遍,合并有机层,有机层浓缩得到12.4g化合物tm1,收率为87.2%。
[0045]
核磁解析:
[0046]
1h-nmr(400mhz,cdcl3):10.39(1h,s),7.64-7.56(1h,m),7.05-7.01(2h,t)。其中,化合物tm1的核磁谱图如图1其所示。
[0047]
1.2化合物a1的合成
[0048]
在氮气保护下,1000ml的三口瓶中加入30.6g化合物sm1(0.1mol),14.9g化合物tm1(0.105mol)和400ml的乙醇溶液,在温度60-70℃条件下,反应5-6h,反应完毕后,将反应液冷却至5-10℃,在此温度条件下慢慢加入4.16g硼氢化钠(0.11mol),升温至40-50℃,在此温度下反应5-6h,反应完毕后,慢慢加入2nhcl调节ph≈7,然后加入300ml dcm,萃取分液,水层用dcm 100ml*2萃取2遍,合并有机层,有机层浓缩干燥得到39.8g的固体化合物a1,
收率为92.3%。
[0049]
核磁解析:
[0050]
1h-nmr(400mhz,cdcl3):8.40-8.37(1h,br),8.28-8.26(2h,m),7.55-7.53(2h,m),7.36-7.30(1h,m),7.00-6.94(2h,m),4.59-4.57(2h,d),4.37-4.31(2h,d),2.44(3h,s),1.42-1.38(3h,t).
[0051]
其中,化合物a1的核磁谱图如图2其所示。
[0052]
1.3化合物b1的合成
[0053]
在氮气保护下,在三口瓶中加入30.6g化合物a1(0.1mol)和甲苯,在0-10℃条件下,慢慢滴加11.88g氯甲酸乙酯(0.11mol),滴加完毕后升温至105-115℃,在此温度下反应3-4h,反应完毕后,降低温度至45-55℃,在此温度条件下滴加5.06g乙醇(0.11mol);然后自然降温到20-25℃,在此温度条件下搅拌1-2h,过滤,滤饼用乙醇洗涤,真空干燥得到48.2g固体化合物b1,收率为95.6%;
[0054]
将所得的产物进行核磁分析,得知该实施例所得的产物即为化合物b1。
[0055]
实施例二:
[0056]
2.1 2,6-二氟苯甲醛的合成
[0057]
在500ml的三口瓶中,加入11.4g(0.1mol)间二氟甲苯,15.1g(0.13mol)tmeda,0.939g(0.007mol)dipa和thf。降低温度至-70~78℃,然后滴加8.27g(0.13mol)丁基锂,滴加完毕后在此温度条件下反应3-6h,然后慢慢滴加10.9g(0.15mol)dmf,在此温度条件下继续反应1-2h,将反应液升到室温,加入饱和的nh4cl溶液,再加入乙酸乙酯,萃取分液,水层用乙酸乙酯洗涤2遍,合并有机层,有机层浓缩得到11.8g化合物tm1,收率为83.6%。
[0058]
2.2化合物a1的合成
[0059]
在氮气保护下,1000ml的三口瓶中加入30.6g化合物sm1(0.1mol),17.0g化合物tm1(0.12mol)和400ml的甲醇溶液,在温度50-60℃条件下,反应2-3h,反应完毕后,将反应液冷却至5-10℃,在此温度条件下慢慢加入8.1g硼氢化钾(0.15mol),升温至50-60℃,在此温度下反应5~6h;反应完毕后,慢慢加入2n hcl,调节ph≈7,然后加入300ml dcm,萃取分液,水层用dcm 100ml*2萃取2遍,合并有机层,有机层浓缩干燥得到36.9g固体化合物a1,收率为85.6%。
[0060]
2.3化合物b1的合成
[0061]
在氮气保护下,在三口瓶中加入30.6g化合物a1(0.1mol)和二氧六环,在0-10℃条件下,慢慢滴加16.2g氯甲酸乙酯(0.15mol),滴加完毕后升温至85-90℃,在此温度下反应8-10h,反应完毕后,降低温度至45-55℃,在此温度条件下滴加6.91g乙醇(0.15mol);然后自然降温到20-25℃,在此温度条件下搅拌1-2h,过滤,滤饼用乙醇洗涤,真空干燥得到45.5g固体化合物b1,收率为90.2%。
[0062]
实施例三:
[0063]
3.1 2,6-二氟苯甲醛的合成
[0064]
在500ml的三口瓶中,加入11.4g(0.1mol)间二氟甲苯,17.5g(0.15mol)tmeda,0.940g(0.007mol)dipa和thf。降低温度至-70~78℃,然后滴加9.5g(0.15mol)丁基锂,滴加完毕后在此温度条件下反应1-2h,然后慢慢滴加12.4g(0.17mol)dmf,在此温度条件下继续反应1-2h,将反应液升到室温,加入饱和的nh4cl溶液,再加入乙酸乙酯,萃取分液,水层
用乙酸乙酯洗涤2遍,合并有机层,有机层浓缩得到11.6gtm1,收率为81.6%。
[0065]
3.2化合物a1的合成
[0066]
在氮气保护,1000ml的三口瓶中加入30.6g化合物sm1(0.1mol),19.88g化合物tm1(0.14mol)和400ml的甲苯溶液,在温度60-70℃条件下,反应10-12h,反应完毕后,将反应液冷却至5-10℃,在此温度条件下慢慢加入4.16g硼氢化钠(0.11mol),升温至70-80℃,在此温度下反应8-9h;反应完毕后,慢慢加入2n hcl,调节ph≈7,然后加入300ml dcm,萃取分液,水层用dcm 100ml*2萃取2遍,合并有机层,有机层浓缩干燥得到34.8g固体化合物a1,收率为80.6%。
[0067]
3.3化合物b1的合成
[0068]
在氮气保护下,在三口瓶中加入30.6g化合物a1(0.1mol)和thf,在0-10℃条件下,慢慢滴加21.6g氯甲酸乙酯(0.2mol),滴加完毕后升温至65-70℃,在此温度下反应12-14h,反应完毕后,降低温度至45-55℃,在此温度条件下滴加9.21g乙醇(0.2mol);然后自然降温到20-25℃,在此温度条件下搅拌1-2h,过滤,滤饼用乙醇洗涤,真空干燥得到41.6g固体化合物b1,收率为82.6%。
[0069]
本发明实施例二和实施例三中化合物的核磁解析数据与实施例一相应化合物的数据基本一致,因此本发明未提供实施例二和实施例三化合物的核磁解析数据及谱图。
[0070]
本发明各原料的上下限取值以及区间值都能实现本发明,以及所列举的各原料都能实现本发明,在此就不一一列举实施例。
[0071]
需要说明的是本发明中提及的所有文献或专利在本技术中引为参考,就如同每一篇文章或者专利被单独因为参考一样。此外应理解以上所述的是本发明的具体实施例及技术原理,在阅读了本发明的上述内容之后,本领域的技术人员可以对本发明作各种改进和改动而不背离本发明的范围,这些等价形式同样落在本发明的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
