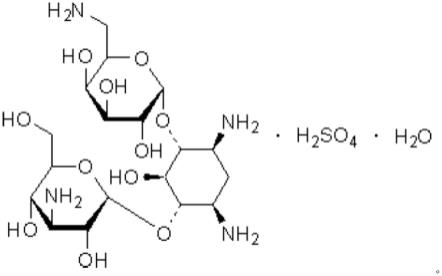
1.本发明属于医药技术领域,涉及制备单硫酸卡那霉素晶体的方法,尤其是涉及制备单硫酸卡那霉素单晶的方法,还涉及该单晶在制药工业中的用途,使用在市售硫酸卡那霉素单硫酸卡那霉素尤其是分子结构中的用途。
背景技术:
2.氨基糖苷类抗生素主要对金黄色葡萄球菌和需氧革兰阴性杆菌,包括铜绿假单胞菌有强大的抗菌作用,对沙雷菌属、布鲁杆菌、沙门杆菌、嗜血杆菌、痢疾杆菌以及结核分支杆菌、其他分支杆菌属亦具有良好的抗菌作用,并且氨基糖苷类抗生素具有较多的优点,如水溶性好、抗菌能力强、性质稳定、吸收排泄较好等,因此在临床上应用较为广泛[蔡焯玲,梁棣昌.氨基糖苷类药物临床应用调查分析[j].深圳中西医结合杂志,2020,30(9):140-141]。
[0003]
卡那霉素(kanamycin)是1958年由日本明治制果药业株式会社开发生产的α-去氧链胺衍生物组成的氨基糖苷类抗菌药,对革兰阴性菌及青霉素、链霉素、红霉素等产生耐药性的金黄色葡萄球菌、大肠埃希菌、产气杆菌、肺炎杆菌和痢疾杆菌有很强的抗菌作用[umezawa h,ueda m,maeda k,et al.production and isolation of a new antibiotic:kanamycin[j].j antibiot(tokyo),1957,10(5):181-188]。目前各国药典均有收录[bp2017、ep10.0、jp17、usp42、cp2020],其原料药根据硫酸盐数目不同分为单硫酸卡那霉素(c
18h36
n4o
11
·
h2so4)和硫酸卡那霉素(c
18h36
n4o
11
·
xh2so4)两种[puius y a,stievater t h,srikrishnan t.crystal structure,conformation,and absolute configuration of kanamycin a[j].carbohyd res,2006,341(17):2871-2875]。
[0004]
如上文所述,卡那霉素成盐后根据硫酸根的数量,分为单硫酸卡那霉素和硫酸卡那霉素,《中国药典》只收载了硫酸卡那霉素(c
18h36
n4o
11
·
xh2so4),未收载单硫酸卡那霉素,但是,国内确实存在单硫酸卡那霉素和硫酸卡那霉素两种原料。硫酸卡那霉素的水溶解度明显高于单硫酸卡那霉素,然而,符合《中国药典》标准的固体原料药硫酸卡那霉素(c
18h36
n4o
11
·
xh2so4)虽然常用于各种制剂的制备,但是这种硫酸卡那霉素(c
18h36
n4o
11
·
xh2so4)具有极强的引湿性,不方便储存和运输,因此一些企业更倾向于生产和使用不具引湿性的单硫酸卡那霉素(c
18h36
n4o
11
·
h2so4)作为固体原料运输和储存,在制剂生产时通过调节硫酸的量来满足制剂配方需求,从这个意义上讲,国内卡那霉素原料药采用其单硫酸盐,将成为一种趋势。
[0005]
《欧洲药典》8.0版(ep8.0)和《英国药典》2016版(bp2016)收载了单硫酸卡那霉素(c
18h36
n4o
11
·
h2so4·
h2o)和硫酸酸卡那霉素,《美国药典》39版(usp39)收载了单硫酸卡那霉素(c
18h36
n4o
11
·
h2so4),《日本药局方》16版(jp16)收载为单硫酸卡那霉素(c
18h36
n4o
11
·
h2so4)和硫酸卡那霉素(c
18h36
n4o
11
·
xh2so4)。
[0006]
根据以上各国药典对单硫酸卡那霉素的收载情况分析,usp39、ep8.0、bp2016和jp16中统一记载单硫酸卡那霉素分子中含有1个硫酸分子(即1:1的状态,在本发明中可简
称为ii态),但是ep8.0和bp2016明确标明单硫酸卡那霉素分子中除含有1个硫酸分子外,还含有1个水分子(即1:1:1的状态,在本发明中可简称为iii态)。
[0007]
那么,国内的单硫酸卡那霉素到底是何种状态,是ii态,还是iii态,和/或,能否为其晶形提供一种简易获得的对照品,等等,仍是本领域技术人员迫切期待的。
技术实现要素:
[0008]
本发明的目的在于以下一个或者多个方面:提供一种单硫酸卡那霉素的单晶,提供一种制备单硫酸卡那霉素的单晶的方法,提供一种验证市售单硫酸卡那霉素的晶形的方法。
[0009]
为此,本发明第一方面提供了一种测定单硫酸卡那霉素杂质含量的方法,其采用hplc-荧光检测法,包括如下步骤:
[0010]
(1)提供hplc-荧光检测法色谱仪器,包括高效液相色谱仪、荧光检测器、柱后衍生化系统;
[0011]
(2)色谱条件:
[0012]
色谱柱:agilent zorbax sb c18柱(4.6mm
×
250mm,5μm);
[0013]
流动相:ph3.4缓冲液(取庚烷磺酸钠一水合物8.7g和无水硫酸钠32g,加水溶解并稀释至成2000ml,用冰醋酸调节ph值至3.4
±
0.1)-甲醇(95:5)为流动相a,以甲醇为流动相b,按下表进行线性梯度洗脱;
[0014]
表:流动相梯度洗脱条件
[0015][0016][0017]
柱温:35℃;
[0018]
流速:1.0ml/min;
[0019]
荧光检测器:激发波长340nm,发射波长455nm;
[0020]
进样体积:5μl;
[0021]
柱后衍生化的衍生化试液:取邻苯二醛0.8g、甲醇300ml、2-巯基乙醇2ml和硼酸缓冲液(取72.0g硼酸和43.0g氢氧化钠,加水溶解并稀释至4000ml,用1mol/l硼酸溶液或1mol/l氢氧化钠溶液调节ph值至10.4
±
0.1)700ml,混合,摇匀,置棕色瓶中,避光储存;
[0022]
衍生化试液流速:0.5ml/min;
[0023]
(3)测定溶液的配制:精密称取供试品适量,加水溶解并定量稀释制成含卡那霉素1mg/ml的溶液,作为供试品溶液;任选地配制卡那霉素对照品溶液、杂质对照品使用卡那霉素b杂质对照品溶液;
[0024]
(4)测定:将供试品溶液和任选的对照品溶液分别注入液相色谱仪,记录色谱图,用峰面积归一化法计算各杂质百分含量和总杂质百分含量。
[0025]
根据本发明第一方面的方法,其中所述单硫酸卡那霉素是具有如下化学结构的一水合物:
[0026][0027]
根据本发明第一方面的方法,其中所述单硫酸卡那霉素为单晶形式。
[0028]
根据本发明第一方面的方法,其中所述单硫酸卡那霉素为单晶形式,其是照包括如下步骤的方法制备得到的:
[0029]
(1)取单硫酸卡那霉素原料药1000mg,置于玻璃管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.5,接着加入1ml异丙醇使混匀;
[0030]
(2)在22~25℃温度条件下将8~12ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0031]
(3)在上述温度条件下静置20~30小时,析出呈柱状的结晶,取出,即得。
[0032]
根据本发明第一方面的方法,其中所述去二氧化碳水是双蒸水煮沸10分钟后冷却至室温所得的水。
[0033]
根据本发明第一方面的方法,其中步骤(1)所述玻璃管是纳氏比色管。
[0034]
根据本发明第一方面的方法,其中步骤(1)中调节溶液的ph值至6.0~6.2。
[0035]
根据本发明第一方面的方法,其中步骤(2)中加10ml甲醇。
[0036]
根据本发明第一方面的方法,其中步骤(3)中静置24小时。
[0037]
根据本发明第一方面的方法,其中所述单硫酸卡那霉素为单晶形式,其是照包括如下步骤的方法制备得到的:
[0038]
(1)取单硫酸卡那霉素原料药1000mg,置于一个50ml的纳氏比色管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.2,接着加入1ml异丙醇使混匀;
[0039]
(2)在22~25℃温度条件下将10ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0040]
(3)在上述温度条件下静置24小时,析出呈柱状的结晶,取出,即得。
[0041]
进一步的,本发明第二方面提供了一种下式化合物的单晶形式:
[0042][0043]
根据本发明第二方面的单晶形式,其为三斜晶系,晶胞参数为根据本发明第二方面的单晶形式,其为三斜晶系,晶胞参数为α=88.48(1)
°
、β=85.21(1)
°
、γ=89.19(1)
°
。
[0044]
根据本发明第二方面的单晶形式,其具有图6(上)所示的粉末x射线衍射图。
[0045]
根据本发明第二方面的单晶形式,其具有图7(下)所示的红外光谱图。
[0046]
根据本发明第二方面的单晶形式,其是照包括如下步骤的方法制备得到的:
[0047]
(1)取单硫酸卡那霉素原料药1000mg,置于玻璃管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.5,接着加入1ml异丙醇使混匀;
[0048]
(2)在22~25℃温度条件下将8~12ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0049]
(3)在上述温度条件下静置20~30小时,析出呈柱状的结晶,取出,即得。
[0050]
根据本发明第二方面的单晶形式,其中所述去二氧化碳水是双蒸水煮沸10分钟后冷却至室温所得的水。
[0051]
根据本发明第二方面的单晶形式,其中步骤(1)所述玻璃管是纳氏比色管。
[0052]
根据本发明第二方面的单晶形式,其中步骤(1)中调节溶液的ph值至6.0~6.2。
[0053]
根据本发明第二方面的单晶形式,其中步骤(2)中加10ml甲醇。
[0054]
根据本发明第二方面的单晶形式,其中步骤(3)中静置24小时。
[0055]
根据本发明第二方面的单晶形式,其中所述单硫酸卡那霉素为单晶形式,其是照包括如下步骤的方法制备得到的:
[0056]
(1)取单硫酸卡那霉素原料药1000mg,置于一个50ml的纳氏比色管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.2,接着加入1ml异丙醇使混匀;
[0057]
(2)在22~25℃温度条件下将10ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0058]
(3)在上述温度条件下静置24小时,析出呈柱状的结晶,取出,即得。
[0059]
进一步的,本发明第三方面提供了一种制备下式化合物的单晶形式的方法:
[0060][0061]
根据本发明第三方面的方法,所述单晶形式为三斜晶系,晶胞参数为根据本发明第三方面的方法,所述单晶形式为三斜晶系,晶胞参数为α=88.48(1)
°
、β=85.21(1)
°
、γ=89.19(1)
°
。
[0062]
根据本发明第三方面的方法,其包括如下步骤:
[0063]
(1)取单硫酸卡那霉素原料药1000mg,置于玻璃管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.5,接着加入1ml异丙醇使混匀;
[0064]
(2)在22~25℃温度条件下将8~12ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0065]
(3)在上述温度条件下静置20~30小时,析出呈柱状的结晶,取出,即得。
[0066]
根据本发明第三方面的方法,其中所述去二氧化碳水是双蒸水煮沸10分钟后冷却至室温所得的水。
[0067]
根据本发明第三方面的方法,其中步骤(1)所述玻璃管是纳氏比色管。
[0068]
根据本发明第三方面的方法,其中步骤(1)中调节溶液的ph值至6.0~6.2。
[0069]
根据本发明第三方面的方法,其中步骤(2)中加10ml甲醇。
[0070]
根据本发明第三方面的方法,其中步骤(3)中静置24小时。
[0071]
根据本发明第三方面的方法,其包括如下步骤:
[0072]
(1)取单硫酸卡那霉素原料药1000mg,置于一个50ml的纳氏比色管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.2,接着加入1ml异丙醇使混匀;
[0073]
(2)在22~25℃温度条件下将10ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0074]
(3)在上述温度条件下静置24小时,析出呈柱状的结晶,取出,即得。
[0075]
进一步的,本发明第四方面提供了本发明第二方面任一项所述单晶形式用于鉴别单硫酸卡那霉素原料药的用途。
[0076]
根据本发明第四方面的用途,其中所述单晶形式具有图6(上)所示的粉末x射线衍射图。
[0077]
根据本发明第四方面的用途,其中所述单晶形式具有图7(下)所示的红外光谱图。
[0078]
在本发明上述各种操作步骤中,虽然其描述的具体步骤在某些细节上或者语言描述上与下文具体实施方式部分的各种实例中所描述的步骤有所区别,然而,本领域技术人员根据本发明全文的详细公开完全可以概括出以上所述方法步骤。
[0079]
本发明的任一方面的任一实施方案,可以与其它实施方案进行组合,只要它们不会出现矛盾。此外,在本发明任一方面的任一实施方案中,任一技术特征可以适用于其它实
施方案中的该技术特征,只要它们不会出现矛盾。下面对本发明作进一步的描述。
[0080]
本发明所引述的所有文献,它们的全部内容通过引用并入本文,并且如果这些文献所表达的含义与本发明不一致时,以本发明的表述为准。此外,本发明使用的各种术语和短语具有本领域技术人员公知的一般含义,即便如此,本发明仍然希望在此对这些术语和短语作更详尽的说明和解释,提及的术语和短语如有与公知含义不一致的,以本发明所表述的含义为准。
[0081]
本发明方法不但能够获得单晶形式的单硫酸卡那霉素一水合物,并且能够显著降低其中杂质的含量。具体地说,实施例1收获的单晶即单硫酸卡那霉素a的收率为78.4%,杂质总量降低4.10倍(5.17%/1.26%)。出人预料的发现是,通过同时控制ph值以及添加异丙醇是降低杂质提高收率所必不可少的条件。例如,在一个补充试验a中,参照实施例1的方法,不同的仅是不添加异丙醇,所得结晶收率为44.7%,杂质总量降低1.56倍(5.17%/3.31%);在另一个补充试验b中,参照实施例1的方法,不同的仅是不调节ph值(步骤(1)原料药加水溶解后的ph=7.14~7.21),所得结晶收率为37.4%,杂质总量降低1.82倍(5.17%/2.84%);在另一个补充试验c中,参照实施例1的方法,不同的仅是不添加异丙醇也不调节ph值(步骤(1)原料药加水溶解后的ph=7.14~7.21),所得结晶收率为38.5%,杂质总量降低1.50倍(5.17%/3.44%)。另外一个补充试验d中,改用m公司批号为190321的单硫酸卡那霉素(照欧洲药典10.0版3030页所载单硫酸卡那霉素方法检测为iii态,其照本发明实施例4之hplc-荧光检测法测定杂质总量5.36%)作为原料药,分别照本发明实施例1、补充试验a、补充试验b、补充试验c制备单晶,结果:用实施例1方法的结晶收率为76.2%、杂质总量降低4.03倍(5.36%/1.33%),用补充试验a方法的结晶收率为46.8%、杂质总量降低1.64倍(5.36%/3.27%),用补充试验b方法的结晶收率为43.2%、杂质总量降低1.72倍(5.36%/3.12%)(步骤(1)原料药加水溶解后的ph=7.19~7.25),用补充试验c方法的结晶收率为47.3%、杂质总量降低1.56倍(5.36%/3.44%)。另外,参照本发明实施例3的方法测定上述补充试验a~d中所得各种结晶产物的粉末x射线衍射图和红外光谱图,结果显示它们与本发明图6(上)粉末衍射图和图7(下)红外光谱图均相同,表明这些改型的补充试验方法结晶收率和杂质总量降低倍数虽然不同但是结晶形式完全相同。
[0082]
本发明呈现如本文各个方面所述优良效果。
附图说明
[0083]
图1:单硫酸卡那霉素绝对构型。
[0084]
图2:单硫酸卡那霉素分子构象。
[0085]
图3:单硫酸卡那霉素晶胞填充图。
[0086]
图4:单硫酸卡那霉素晶胞内氢键盐键排布图。
[0087]
图5:单硫酸卡那霉素结晶的电子显微镜晶型图。
[0088]
图6:单硫酸卡那霉素结晶前后粉末x射线衍射对比图。
[0089]
图7:单硫酸卡那霉素原料和结晶后红外光谱对比图。
[0090]
图8:单硫酸卡那霉素结晶前后的hplc色谱图。
具体实施方式
[0091]
通过下面的实施例可以对本发明进行进一步的描述,然而,本发明的范围并不限于下述实施例。本领域的专业人员能够理解,在不背离本发明的精神和范围的前提下,可以对本发明进行各种变化和修饰。本发明对试验中所使用到的材料以及试验方法进行一般性和/或具体的描述。虽然为实现本发明目的所使用的许多材料和操作方法是本领域公知的,但是本发明仍然在此作尽可能详细描述。以下实施例进一步说明本发明,而不是限制本发明。以下实施例所用的单硫酸卡那霉素的原料药,为市售品并经测定为单硫酸卡那霉素一水合物(iii态,照欧洲药典10.0版3030页所载单硫酸卡那霉素方法检测,b公司批号为201201的产品)。
[0092]
实施例1、制备单硫酸卡那霉素的单晶
[0093]
本实施例制备单硫酸卡那霉素的单晶,操作步骤如下:
[0094]
(1)取单硫酸卡那霉素原料药1000mg,置于一个50ml的纳氏比色管中,加入20ml去二氧化碳水使溶解,测定ph值并用1m硫酸溶液调节溶液的ph值至6.0~6.2,接着加入1ml异丙醇使混匀;
[0095]
(2)在22~25℃温度条件下将10ml甲醇以2ml/min的速度缓缓从步骤(1)所得溶液的液面加入;
[0096]
(3)在上述温度条件下静置24小时,析出呈柱状的结晶,取出,即得。所得结晶在本发明中可称为单硫酸卡那霉素a。
[0097]
所用去二氧化碳水是双蒸水煮沸10分钟后冷却至室温所得的水。
[0098]
实施例2、单晶x射线衍射(xrd)测定单硫酸卡那霉素晶体
[0099]
本实施例使用单晶x射线衍射(xrd)仪,测定实施例1所得的单硫酸卡那霉素的柱状结晶,以证明其晶体结构。
[0100]
根据各国药典对单硫酸卡那霉素的收载情况分析,其中包括chp2000版、usp39、ep8.0、bp2016和jp16记载关于单硫酸卡那霉素分子式中水分子的差异问题,本实施例通过对培养获得的单硫酸卡那霉素结晶利用单晶x射线衍射仪进行测定,验证所得到的单硫酸卡那霉素晶体的分子式,并且测定了单硫酸卡那霉素晶体的绝对构型、构象、晶胞、晶胞内氢键与盐键排布。
[0101]
样品:实施例1所得的单硫酸卡那霉素的柱状结晶,结晶长度大于0.4mm。
[0102]
仪器:bruker smart apex-ii型单晶x射线衍射仪。
[0103]
测定条件和晶体结构数据结果如下表1。
[0104]
表1:单硫酸卡那霉素晶体结构数据结果
[0105][0106]
根据上述结果,本实施例测得了实施例1所得单硫酸卡那霉素的化学计量式、分子量、所属晶系、空间群、晶胞中分子数z、晶胞体积、晶体密度、晶胞参数(a,b,c;α,β,γ),另外包括总标度因子f(000)、基于全矩阵最小二乘法的最终可靠因子、残差因子r1、基于f0的加权残差因子wr2与拟合优度s。
[0107]
图1显示了单硫酸卡那霉素绝对构型,图2显示了单硫酸卡那霉素分子构象,图3显示了单硫酸卡那霉素晶胞填充图,根据晶胞填充图证实了实施例1所得单硫酸卡那霉素晶体的单晶结构。
[0108]
另外,本实施例2所测得的晶胞氢键和盐键见表2。
[0109]
表2:单硫酸卡那霉素晶胞氢键和盐键结果
[0110][0111]
获得的单硫酸卡那霉素晶胞内氢键盐键排布图见图4所示。
[0112]
根据本实施例所述各表格和附图关于单硫酸卡那霉素晶体数据测定结果,充分证明了实施例1所得单硫酸卡那霉素的分子式为c
18h36
n4o
11
·
h2so4·
h2o,确定了单硫酸卡那霉素的绝对构型、分子构象和晶胞结构,通过单硫酸卡那霉素的绝对构型、单硫酸卡那霉素分子构型、单硫酸卡那霉素的晶胞填充图、晶胞氢键和盐键结果、单硫酸卡那霉素晶胞内氢键盐键排布图,证明了单硫酸卡那霉素分子内硫酸分子和水分子的结合方式。本实施例利用单晶x射线衍射实验,不仅证实了单硫酸卡那霉素分子式,更反映出了单硫酸卡那霉素晶体内在和外在存在的总体状态。
[0113]
本实施例2测定实施例1所得单硫酸卡那霉素单晶的晶体结构数据,涉及的原子坐标参数见表3。
[0114]
表3:单硫酸卡那霉素单晶原子坐标参数表
[0115]
[0116]
[0117][0118]
本实施例2测定实施例1所得单硫酸卡那霉素单晶的晶体结构数据,涉及的原子等价温度因子值见表4。
[0119]
表4:原子等价温度因子值表
[0120]
[0121][0122]
本实施例2测定实施例1所得单硫酸卡那霉素单晶的晶体结构数据,涉及的成键原子键长值见表5。
[0123]
表5:成键原子键长表
[0124]
[0125][0126]
本实施例2测定实施例1所得单硫酸卡那霉素单晶的晶体结构数据,涉及的成键原子键角值见表6。
[0127]
表6:成键原子键角值表
[0128]
[0129]
[0130]
[0131][0132]
实施例3、单硫酸卡那霉素单晶与单硫酸卡那霉素原料晶体一致性证明
[0133]
本实施例对实施例1所得的单硫酸卡那霉素结晶(单硫酸卡那霉素a,单晶)与所用的单硫酸卡那霉素原料药进行晶体的一致性证明。
[0134]
1、电子显微镜
[0135]
采用光学偏光显微镜进行初步检查,证明市售单硫酸卡那霉素原料药具有结晶性,经过实施例1重结晶后,得到新的晶体(单硫酸卡那霉素a),用日立s-4800冷场发射扫描电子显微镜下观察晶体的性状,结果详见图5之单硫酸卡那霉素的电子显微镜晶型图。
[0136]
2、粉末x射线衍射
[0137]
为了进一步证实单硫酸卡那霉素的结晶性,通过粉末x射线衍射光谱比较,单硫酸卡那霉素原料和重结晶后的单硫酸卡那霉素的粉末x射线衍射图谱无差异,结果详见图6。
[0138]
3、红外光谱
[0139]
另外也对比了单硫酸卡那霉素的红外光谱图,详见图7单硫酸卡那霉素原料和结晶后红外光谱对比图,单硫酸卡那霉素原料和结晶后无明显区别,表明重结晶条件并没有改变晶体结构。
[0140]
根据电子显微观察结果,并且结合粉末x射线衍射图谱,证实了单硫酸卡那霉素原料具有结晶性。
[0141]
原料重结晶的目的主要是培养更加完整的晶型,有利于单晶x射线衍射测定单硫酸卡那霉素的分子式,从粉末x射线衍射谱图和红外光谱图也证实了晶型培养的可靠性。
[0142]
实施例4、单硫酸卡那霉素单晶与单硫酸卡那霉素原料的化学纯度测定
[0143]
本实施例使用hplc-荧光检测法,对实施例1所得的单硫酸卡那霉素结晶(单硫酸卡那霉素a,单晶)与所用的单硫酸卡那霉素原料药进行化学纯度测定。
[0144]
1、hplc-荧光检测法色谱条件
[0145]
所用的部分仪器:lc-20a高效液相色谱仪(岛津);rf-20a荧光检测器(岛津);pickering pcx 5200柱后衍生化系统(沃特斯)。
[0146]
所用的部分试剂:硫酸卡那霉素对照品(批号130556-201502,中国食品药品检定研究院);卡那霉素b对照品(批号:130548-200501,中国食品药品检定研究院);单硫酸卡那霉素样品b(b公司,批号:201201)。
[0147]
色谱柱:agilent zorbax sb c18柱(4.6mm
×
250mm,5μm);流动相:ph3.4缓冲液(取庚烷磺酸钠一水合物8.7g和无水硫酸钠32g,加水溶解并稀释至成2000ml,用冰醋酸调节ph值至3.4
±
0.1)-甲醇(95:5)为流动相a,以甲醇为流动相b,按表7进行线性梯度洗脱;柱温:35℃;流速:1.0ml/min;荧光检测器:激发波长340nm,发射波长455nm;进样体积:5μl。配有柱后衍生化装置,衍生化试液:取邻苯二醛0.8g、甲醇300ml、2-巯基乙醇2ml和硼酸缓冲液(取72.0g硼酸和43.0g氢氧化钠,加水溶解并稀释至4000ml,用1mol/l硼酸溶液或
1mol/l氢氧化钠溶液调节ph值至10.4
±
0.1)700ml,混合,摇匀,置棕色瓶中,避光储存;衍生化试液流速:0.5ml/min。
[0148]
表7:流动相梯度洗脱条件
[0149]
t/mina/%b/%09010108020208020306040406040419010519010
[0150]
有关物质测定溶液:精密称取供试品适量,加水溶解并定量稀释制成约含卡那霉素1mg/ml的溶液,作为有关物质供试品溶液。
[0151]
采用峰面积归一法计算有关物质杂质的含量,单硫酸卡那霉素结晶前后的hplc图谱见图8,其中显示4~17min期间的杂质显著减少甚至消失。另外,如图8所示,原料药显示有6个相当显著的杂质,各杂质之间的分离度均大于4.0(t=13.287min与t=13.543min二者之间分离度最小为4.26),显示优良的分离度。
[0152]
经测定并计算,实施例1所得单硫酸卡那霉素结晶(单硫酸卡那霉素a,单晶)的杂质总量(%,峰面积归一化法)为1.26%,制备该单晶所用单硫酸卡那霉素原料药的杂质总量(%,峰面积归一化法)为5.17%,杂质总量降低4.10倍(5.17%/1.26%)。
[0153]
本发明还照《中国药典2020年版》二部1567页硫酸卡那霉素品种关于测定卡那霉素b项下的方法,测定同批原料药,结果仅显示有3个杂质并且归一化法计算为5.52%,表明该药典方法的可靠性明显比本发明方法差。
[0154]
研究结果表明,通过结晶后的单硫酸卡那霉素杂质含量明显降低,质量明显优于市场上的任何硫酸卡那霉素原料的质量。
[0155]
以上所述实施例仅是为充分说明本技术而所举的较佳的实施例,本技术的保护范围不限于此。本技术领域的技术人员在本技术基础上所作的等同替代或变换,均在本技术的保护范围之内。本技术的保护范围以权利要求书为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。