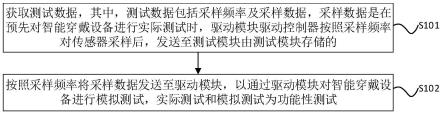
一种手性cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用
技术领域
1.本发明涉及手性拆分技术领域,尤其涉及一种手性cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用。
背景技术:
2.手性药物是指药物分子结构中引入手性中心后,得到的一对互为实物与镜像的对映异构体,每一对化学纯的对映异构体的理化性质有所不同,显示出不同的药理学、毒理学以及药代动力学性质,手性药物的不同异构体在生物体内吸收、分布、代谢和排泄均体现出立体选择性。
3.由于特定的杂环结构和药理功效,二氢嘧啶酮(dhpms)及其衍生物具有抗癌活性、抗菌活性、抗氧化、抗炎、钙离子通道抑制剂、抗高血压等。这类化合物因其具备广泛多样的生物活性,促进了各种对抗疾病新药的大量研究,例如sq 32926、sq 32547、snap 6201、monastrol、bay 41-4109等。研究发现,dhpms的分子结构中4-位手性中心的绝对构型与其生物活性有着密切联系,单一的对映体往往表现出不同甚至相反的药理活性,例如,钠离子通道阻断剂r-sq32926体外测试其降压活性是其对映体的400多倍;有丝分裂驱动蛋白eg5抑制剂s-monastrol的药效是r-monastrol的15倍;(s)-l-771688作为α1a受体拮抗剂,可用于良性前列腺增生的治疗,其药效强于其对映体。因此,手性药物的拆分对于药品的安全使用具有非常重要的意义。
4.目前,具有均一手性元素、永久孔隙率的多样化结构的手性金属-有机骨架(mofs)材料作为手性固定相(csp)在拆分对映体方面受到广泛关注。因此,如何提供一种具有高效拆分能力的mofs材料对dhpms的手性拆分在生物制药、药物代谢动力学以及药理学等研究中具有重要意义。
技术实现要素:
5.本发明的目的在于提供一种手性cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,解决上述现有技术存在的问题。
6.为了实现上述发明目的,本发明提供以下技术方案:
7.本发明提供了一种手性cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,利用高效液相色谱柱拆分二氢嘧啶酮衍生物;所述高效液相色谱柱的固定相填料为手性cd-mofs材料;
8.其中,所述手性cd-mofs材料的无机金属中心为镉离子,有机配体为手性羧酸配体和4,4'-联吡啶。
9.优选的,在上述应用中,所述高效液相色谱柱的制备方法包括以下步骤:
10.将手性cd-mofs材料填装于不锈钢柱中,得到高效液相色谱柱。
11.优选的,在上述应用中,所述手性cd-mofs材料的粒径为10~10.5μm。
12.优选的,在上述应用中,所述填装的压力为38~42mpa。
13.优选的,在上述应用中,所述手性cd-mofs材料中,手性羧酸配体的结构式为以下结构式中的一种:
[0014][0015]
优选的,在上述应用中,所述手性cd-mofs材料中,镉离子为七配位,包括六个来自手性羧酸配体的氧原子和一个来自4,4'-联吡啶配体的氮原子;所述手性羧酸配体、4,4'-联吡啶和镉离子的摩尔比为1:0.5~2:0.5~4。
[0016]
优选的,在上述应用中,所述利用高效液相色谱柱拆分二氢嘧啶酮衍生物的色谱条件为:
[0017]
流动相为正己烷和异丙醇,流动相流速为0.5~2.0ml/min,色谱柱温度为20~50℃,检测波长为220~260nm。
[0018]
优选的,在上述应用中,所述流动相中正己烷和异丙醇的体积比为80~95:5~15。
[0019]
经由上述的技术方案可知,与现有技术相比,本发明具有如下有益效果:
[0020]
本发明的手性cd-mofs材料拆分二氢嘧啶酮衍生物的原理是通过构建手性材料和固定相-拆分物之间强氢键的结合作用,利用手性材料结构高比表面积和材料本身立体选择性来实现二氢嘧啶酮对映体的分离。该材料可用于高效液相色谱柱的固定相填料,拓宽了手性材料的应用范围;而且,在当前拆分条件下,该材料对二氢嘧啶酮对映体的拆分分离因子和分离度大,结果显示良好的手性拆分性能;同时,最优拆分条件可以根据实际分离要求与分离条件灵活调节。
附图说明
[0021]
为了更清楚地说明本发明实施例或现有技术中的技术方案,以下将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
[0022]
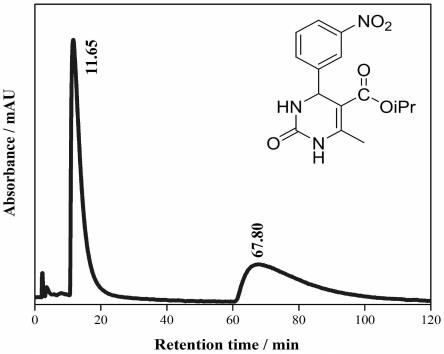
图1为实施例1的二氢嘧啶酮衍生物6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯的高效液相色谱分离结果;
[0023]
图2为实施例2的二氢嘧啶酮衍生物6-甲基-2-氧代-4-苯基-1,2,3,4-四氢嘧啶-5-羧酸乙酯的高效液相色谱分离结果;
[0024]
图3为实施例3的二氢嘧啶酮衍生物4-(4-硝基苯基)-6-甲基-2-氧代-1,2,3,4-四氢嘧啶-5-甲酸乙酯的高效液相色谱分离结果;
[0025]
图4为实施例4的二氢嘧啶酮衍生物4-(4-叔丁基苯基)-6-甲基-2-氧代-1,2,3,4-四氢嘧啶-5-甲酸乙酯的高效液相色谱分离结果;
[0026]
图5为实施例1的手性r-cd-mofs材料的不对称单元图。
具体实施方式
[0027]
本发明提供一种手性cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,利用高效液相色谱柱拆分二氢嘧啶酮衍生物;
[0028]
其中,高效液相色谱柱的固定相填料为手性cd-mofs材料;
[0029]
手性cd-mofs材料的无机金属中心为镉离子,有机配体为手性羧酸配体和4,4'-联吡啶。
[0030]
在本发明中,高效液相色谱柱的制备方法包括以下步骤:
[0031]
将手性cd-mofs材料填装于不锈钢柱中,得到高效液相色谱柱。
[0032]
在本发明中,手性cd-mofs材料的粒径优选为10~10.5μm,进一步优选为10.1~10.4μm,更优选为10.3μm。
[0033]
在本发明中,填装优选为高压匀浆法;填装的压力优选为38~42mpa,进一步优选为39~41mpa,更优选为40mpa;高压匀浆法的匀浆液优选为异丙醇,顶替液优选为异丙醇和正己烷;异丙醇和正己烷的体积比优选为10:90。
[0034]
在本发明中,手性cd-mofs材料中,手性羧酸配体的结构式优选为以下结构式中的一种:
[0035][0036]
在本发明中,手性cd-mofs材料中,镉离子为七配位,包括六个来自手性羧酸配体的氧原子和一个来自4,4'-联吡啶配体的氮原子;其中,一个手性羧酸配体中的一个羧酸端基中的两个氧原子与同一镉离子配位。本发明的材料通过手性羧酸主配体将镉离子连接成一维链,再通过4,4'-联吡啶将其连接为二维网状金属框架有机材料。
[0037]
在本发明中,手性羧酸配体、4,4'-联吡啶和镉离子的摩尔比优选为1:0.5~2:0.5~4,进一步优选为1:0.7~1.8:0.6~3.5,更优选为1:1.1:2.3。
[0038]
在本发明中,利用高效液相色谱柱拆分二氢嘧啶酮衍生物的色谱条件为:
[0039]
流动相优选为正己烷和异丙醇;流动相流速优选为0.5~2.0ml/min,进一步优选为0.8~1.7ml/min,更优选为1.3ml/min;色谱柱温度优选为20~50℃,进一步优选为22~42℃,更优选为36℃;检测波长优选为220~260nm,进一步优选为226~259nm,更优选为248nm。
[0040]
在本发明中,流动相中正己烷和异丙醇的体积比优选为80~95:5~15,进一步优选为82~91:7~13,更优选为89:11。
[0041]
本发明还提供上述手性cd-mofs材料的制备方法,包括以下步骤:
[0042]
(1)中间产物的制备:将1,2-环己烷二胺、三乙胺和无水二氯甲烷混合,然后加入草酰氯单乙酯的无水二氯甲烷溶液进行反应,反应结束后将产物萃取、干燥、纯化,得到中间产物;
[0043]
(2)手性羧酸配体的制备:将中间产物溶于乙醇和水的混合溶液中,再加入氢氧化钠,加热进行反应,反应结束后调节ph≤1,减压旋蒸得到产物,将产物过滤、洗涤、干燥,得到手性羧酸配体;
[0044]
(3)手性cd-mofs材料的制备:将手性羧酸配体、4,4'-联吡啶溶于n,n-二甲基甲酰胺中,再加入四水硝酸镉和水,加热反应,反应结束后将产物过滤、洗涤、干燥,得到手性cd-mofs材料。
[0045]
在本发明中,步骤(1)中1,2-环己烷二胺优选为(1r,2r)-环己烷-1,2-二胺或(1s,2s)-环己烷-1,2-二胺,进一步优选为(1r,2r)-环己烷-1,2-二胺。
[0046]
在本发明中,步骤(1)中1,2-环己烷二胺、三乙胺和草酰氯单乙酯的摩尔比优选为1:1~5:1~6,进一步优选为1:1.3~4.6:2~5,更优选为1:2.7:3。
[0047]
在本发明中,步骤(1)中无水二氯甲烷和草酰氯单乙酯的无水二氯甲烷溶液的体积比优选为4~6:1,进一步优选为4.3~5.7:1,更优选为4.9:1。
[0048]
在本发明中,步骤(1)中反应的保护气体优选为氩气;反应的温度优选为0~5℃,进一步优选为1~4℃,更优选为2℃;反应的时间优选为6~12h,进一步优选为7~10h,更优选为8h。
[0049]
在本发明中,步骤(1)纯化优选为硅胶柱纯化;纯化的洗脱剂优选为石油醚和乙酸乙酯;石油醚和乙酸乙酯的体积比优选为3:1。
[0050]
在本发明中,步骤(2)中乙醇和水的体积比优选为1:1。
[0051]
在本发明中,步骤(2)中中间产物和混合溶液的质量体积比优选为3~10g:50~80ml,进一步优选为4~9g:55~76ml,更优选为6g:60ml。
[0052]
在本发明中,步骤(2)中中间产物和氢氧化钠的摩尔比优选为1:1~5,进一步优选为1:2~4,更优选为1:2.5。
[0053]
在本发明中,步骤(2)中反应的温度优选为85~95℃,进一步优选为87~93℃,更优选为89℃;反应的时间优选为10~14h,进一步优选为11~13h,更优选为12h。
[0054]
在本发明中,步骤(3)中手性羧酸配体、4,4'-联吡啶和四水硝酸镉的摩尔比优选为1:0.5~2:0.5~4,进一步优选为1:0.7~1.6:1.1~3.2,更优选为1:1.2:2.5。
[0055]
在本发明中,步骤(3)中手性羧酸配体、n,n-二甲基甲酰胺和水的摩尔体积比优选为1~3mmol:100ml:400~500ml,进一步优选为1.2~2.6mmol:100ml:420~490ml,更优选为2.3mmol:100ml:470ml。
[0056]
在本发明中,步骤(3)中反应的温度优选为80~120℃,进一步优选为87~114℃,更优选为96℃;反应的时间优选为1~5天,进一步优选为1.2~4.5天,更优选为3天。
[0057]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0058]
实施例1
[0059]
本实施例提供一种手性r-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体为:
[0060]
通过高压匀浆法,将手性r-cd-mofs材料(粒径为10μm)在40mpa压力下填装于不锈
钢柱(150mm long
×
4.6mm i.d.)中,得到高效液相色谱柱;
[0061]
将高效液相色谱柱使用体积比为90:10的正己烷和异丙醇冲洗和平衡,直到基线稳定;然后进行色谱分离,色谱条件为:以1mg/ml的6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯(记为二氢嘧啶酮衍生物)作为待测液,以体积比为90:10的正己烷和异丙醇作为流动相,以1.0ml/min的流速分离待测液中的二氢嘧啶酮衍生物,色谱柱温度为20℃,使用紫外检测器在254nm的检测波长下检测紫外信号;
[0062]
色谱分离结束后,使用体积比为90:10的正己烷和异丙醇洗脱色谱柱0.5h,然后将色谱柱用螺丝堵头封端,并在室温下储存。
[0063]
其中,手性r-cd-mofs材料的制备方法包括以下步骤:
[0064]
(1)手性羧酸配体r-h2l的制备如下所示:
[0065][0066]
在氩气保护下,将(1r,2r)-环己烷-1,2-二胺(5.709g,0.05mol)、三乙胺(21ml,0.15mol)用100ml无水二氯甲烷溶解成无色透明的混合溶液;再逐滴滴加20ml含有草酰氯单乙酯(14.336g,0.105mol)的无水二氯甲烷溶液;滴加完毕后于0℃搅拌反应8h,得到淡黄色固液混合物;用150ml饱和nahco3溶液猝灭反应后,用二氯甲烷萃取分离得到黄色有机相,用无水mgso4干燥有机相后拌硅胶旋干,得到的粗产物通过硅胶柱纯化(洗脱剂为体积比为3:1的pe和ea),得到中间产物r-1;
[0067]
将中间产物r-1(6.283g,0.02mol)溶于50ml乙醇和水体积比为1:1的混合溶液中,再加入氢氧化钠(2g,0.05mol),加热至90℃反应12h,反应结束后使用盐酸调节ph≤1,减压旋蒸得到产物,将产物过滤、用水洗涤5次、干燥,得到手性羧酸配体r-h2l(3.215g,产率为63%);
[0068]
(2)手性r-cd-mofs材料的制备:将手性羧酸配体r-h2l(25.8mg,0.01mmol)、4,4'-联吡啶(15.6mg,0.01mmol)溶于1mln,n-二甲基甲酰胺中,再加入四水硝酸镉(30.8mg,0.01mmol)和5ml水,加热至90℃反应36h,反应结束后将产物过滤、乙醇洗涤5次、干燥,得到手性r-cd-mofs材料(产率为35%)。
[0069]
上述手性r-cd-mofs材料的不对称单元图如图5所示。由图5可知,手性r-cd-mofs材料中镉离子为七配位,包含六个来自手性羧酸配体的氧原子以及一个4,4'-联吡啶辅助配体的氮原子,其中,一个手性羧酸配体中的一个羧酸端基中的两个氧原子与同一镉离子配位。
[0070]
上述二氢嘧啶酮衍生物6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯的高效液相色谱分离结果如图1所示。由图1可知,6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯的液相色谱拆分结果显示对映体基线分离,实现了对消旋体良好的拆分,拆分数据分析为k1=4.18,k2=29.16,分离因子α=6.97,分离度rs=3.01。
[0071]
实施例2
[0072]
本实施例提供一种手性r-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体参见实施例1,不同之处在于将实施例1的6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯替换为6-甲基-2-氧代-4-苯基-1,2,3,4-四氢嘧啶-5-羧酸乙酯。
[0073]
上述二氢嘧啶酮衍生物6-甲基-2-氧代-4-苯基-1,2,3,4-四氢嘧啶-5-羧酸乙酯的高效液相色谱分离结果如图2所示。由图2可知,两个对映体基本基线分离,实现了对消旋体良好的拆分,拆分数据分析为k1=3.72,k2=14.62,分离因子α=3.93,分离度rs=1.50。
[0074]
实施例3
[0075]
本实施例提供一种手性r-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体参见实施例1,不同之处在于将实施例1的6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯替换为4-(4-硝基苯基)-6-甲基-2-氧代-1,2,3,4-四氢嘧啶-5-甲酸乙酯。
[0076]
上述二氢嘧啶酮衍生物4-(4-硝基苯基)-6-甲基-2-氧代-1,2,3,4-四氢嘧啶-5-甲酸乙酯的高效液相色谱分离结果如图3所示。由图3可知,两个对映体基线分离,实现了对消旋体良好的拆分。拆分数据分析为k1=2.43,k2=11.69,分离因子α=4.81,分离度rs=1.85。
[0077]
实施例4
[0078]
本实施例提供一种手性r-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体参见实施例1,不同之处在于将实施例1的6-甲基-4-(3-硝基苯基)-2-氧代-1,2,3,4-四氢嘧啶-5-羧酸异丙酯替换为4-(4-叔丁基苯基)-6-甲基-2-氧代-1,2,3,4-四氢嘧啶-5-甲酸乙酯。
[0079]
上述二氢嘧啶酮衍生物4-(4-叔丁基苯基)-6-甲基-2-氧代-1,2,3,4-四氢嘧啶-5-甲酸乙酯的高效液相色谱分离结果如图4所示。由图4可知,两个对映体基线分离,实现了对消旋体良好的拆分。拆分数据分析为k1=0.53,k2=2.07,分离因子α=3.87,分离度rs=1.46。
[0080]
实施例5
[0081]
本实施例提供一种手性s-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体参见实施例1,不同之处在于高效液相色谱柱的固定相填料为手性s-cd-mofs材料;手性s-cd-mofs材料的制备方法参见实施例1,不同之处在于将步骤(1)中的(1r,2r)-环己烷-1,2-二胺替换为(1s,2s)-环己烷-1,2-二胺。
[0082]
实施例6
[0083]
本实施例提供一种手性r-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体参见实施例1,不同之处在于手性r-cd-mofs材料的粒径为10.5μm。
[0084]
实施例7
[0085]
本实施例提供一种手性r-cd-mofs材料在高效液相色谱中拆分二氢嘧啶酮衍生物的应用,具体参见实施例1,不同之处在于手性r-cd-mofs材料的制备方法中,步骤(2)中手性羧酸配体r-h2l为0.01mmol,4,4'-联吡啶为0.02mmol,四水硝酸镉为0.03mmol。
[0086]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人
员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。