1.本发明涉及计算机辅助药物设计技术以及生物和药物信息学领域,尤其涉及一种靶标-配体结合亲和力预测方法。
背景技术:
2.大多数生物过程由生物分子识别决定,其中蛋白质通常作为靶标与配体相互作用以调节生物功能,例如酶催化,信号转导等。靶标-配体相互作用的研究是重要的课题。靶标-配体相互作用的结合强度(以实数衡量)的数量通常定义为结合亲和力,可以通过抑制常数ki、解离常数kd和半数最大抑制浓度ic
50
来量化。现有药物大部分都是具有生物活性的小分子化合物,识别与靶蛋白具有高亲和力的配体(小分子候选药物)是药物发现早期的一项主要任务。
3.实验试错方法确定靶标-配体的亲和力存在耗时费力的问题。基于半经验的3d-定量构效关系模型虽预测靶标-配体亲和力的准确度高,且解释性强,但其通用性差,仅适用于某一类骨架的分子。基于物理驱动的方法(如分子动力学方法)具有较高的准确性和良好的解释性,然而巨大的计算时间成本阻碍了其在高通量筛选中的应用。相比于分子动力学方法,分子对接方法可大幅降低计算时间成本,但也牺牲了较多精度。基于数据驱动的方法(如机器学习方法)计算速度极快,然而大多数机器学习算法仍依赖专家知识进行特征提取/选择,在大数据集中难以权衡准确性和通用性。在海量数据和强大的并行计算能力推动下,由传统机器学习方法进一步发展得到的深度学习方法具有更强的数据拟合能力,尽管目前已提出了许多深度学习模型来预测结合亲和力,但大多数需要靶标-配体复合物的3d结构特征,与文本特征相比,3d结构特征不容易获得。
技术实现要素:
4.针对现有技术存在的上述问题,本发明的目的是充分利用文本特征的优势,开发一种端到端的基于门控注意力机制的深度学习模型,以快速识别具有高结合亲和力的靶标-配体复合物。
5.为实现上述目的,本发明采用如下技术方案,包括以下具体步骤:
6.基于门控注意力机制的靶标-配体结合亲和力的深度学习预测方法,包括以下具体步骤:
7.步骤1:建立结合亲和力数据库,用于后续的深度学习模型。
8.步骤2:将配体的smiles字符串转换为配体矩阵,作为深度学习模型的输入。
9.步骤3:将蛋白质的氨基酸序列转换为蛋白质矩阵,作为深度学习模型的输入。
10.步骤4:构建用于模型训练的深度学习模型的架构,将配体矩阵送到一个全连接层和一个基于门增强的注意力层用于特征提取,通过矩阵行的加和方式来聚合配体片段的特征向量。
11.步骤5:将蛋白质矩阵先送到一维卷积层和最大池化层,以减少蛋白质矩阵的行数,然后再送入基于门增强的注意力层用于特征提取,通过矩阵行的加和方式来聚合蛋白质高阶氨基酸的特征向量。
12.步骤6:将聚合后的配体片段的特征向量与蛋白质高阶氨基酸的特征向量拼接在一起送入后续的全连接层以预测靶标-配体复合物的高/低结合亲和力的概率。
13.进一步地,步骤1具体包括:根据以下标准对结合亲和力数据库进行预处理:
14.步骤1.1:删除涉及ic
50
性质的条目,保留涉及ki和kd性质的条目。
15.步骤1.2:删除rdkit工具读取配体结构文件失败或mol2vec方法找不到配体morgen片段的条目。
16.步骤1.3:分别删除morgen片段或氨基酸数大于阈值的靶标-配体复合物条目。
17.步骤1.4:k
i/d
《n的靶标-配体复合物标记为高结合亲和力,用“1”表示,而k
i/d
≥n的靶标-配体复合物标记为低结合亲和力,用“0”表示,k
i/d
表示ki或kd,n表示抑制或解离常数阈值。
18.最后,在应用上述步骤1.1-1.4标准后,获得独特的靶标-配体复合物样本存储在结合亲和力数据库中,用于后续的深度学习模型。
19.进一步地,步骤2具体包括:
20.步骤2.1:使用morgan算法将分子的smiles字符串表示转换为morgen片段。
21.步骤2.2:将mol2vec中的morgen片段的特征向量嵌入到步骤2.1得到的morgen片段中。将每个分子的morgen片段的特征向量求和来表示整个分子的特征向量。
22.步骤2.3:基于mol2vec描述符,每个配体样本由一个二维矩阵表示,其中矩阵行表示morgen片段,矩阵列表示片段的特征向量。
23.进一步地,步骤3具体包括:
24.步骤3.1:将蛋白质序列中每三个相邻的氨基酸视为一个高阶氨基酸。通过这种方式,将结合亲和力数据库中的蛋白质编写成高阶氨基酸字符串。
25.步骤3.2:通过深度学习模型中的嵌入方法,给每个高阶氨基酸字符串嵌入一个特征向量。
26.步骤3.3:每个蛋白质样本由一个二维矩阵表示,其中,矩阵行表示高阶氨基酸字符串,矩阵列表示高阶氨基酸字符串的特征向量。
27.进一步地,步骤4具体包括:
28.步骤4.1:将配体矩阵x
l
送到全连接层v以将配体特征的维数降低。
29.步骤4.2:将获得的降维后的配体矩阵x
′
l
送到基于门增强的注意力层。在注意力层中,x
′
l
首先通过一个可学习的权重矩阵w1进行变换,以获得具有显著特征的新配体矩阵x
″
l
。
30.步骤4.3:通过方程得到注意力系数e
ij
,其中e
ij
表示第j个片段特征对第i个片段特征的重要性,通过求和和来强制e
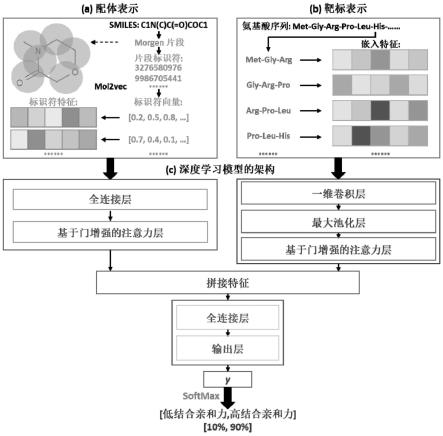
ij
等于e
ji
。
31.步骤4.4:将e
ij
送到一个softmax激活函数,通过a
ij
=exp(e
ij
)/∑jexp(e
ij
)等式得到归一化的注意力系数a
ij
。
32.步骤4.5:利用得到的a
ij
,通过x
″′
l,i
=∑
jaij
x
″
l,j
等式将每个片段特征更新为相邻
片段特征的线性组合,并且随后将relu激活函数添加到x
″′
l,i
。
33.步骤4.6:应用门增强算法,通过前一个片段特征x
′
l,i
和x
″′
l,i
的线性组合使用方程来获得基于门增强的注意力层的输出片段特征zi表示为zi=σ((x
′
l,i
||x
″′
l,i
)u)并解释为x
′
l,i
和x
″′
l,i
传递给的权重系数,其中u是可学习向量,σ表示sigmoid激活函数,(
·
||
·
)是拼接两个向量的操作符。
34.步骤4.7:通过等式,将配体片段的特征向量相加成一个表示配体特征的向量。
35.进一步地,步骤5具体包括:
36.步骤5.1:将蛋白质矩阵x
p
送到一维卷积层和最大池化层。通过这种方式,一维卷积层和最大池化层能够从整个蛋白质序列中提取关键的高阶氨基酸,这有助于将氨基酸特征集中在靶标-配体复合物的口袋区域,同时减少模型冗余并节省下一个注意力层的计算时间成本。
37.步骤5.2:将步骤5.1处理的蛋白质矩阵x
′
p
送到基于门增强的注意力层。在注意力层中,x
′
p
首先通过一个可学习的权重矩阵w2进行变换,以获得具有显著特征的新蛋白质矩阵x
″
p
。
38.步骤5.3:通过方程得到注意力系数e
kl
,其中e
kl
表示第l个高阶氨基酸特征对第k个高阶氨基酸特征的重要性,通过求和和来强制e
kl
等于e
lk
。
39.步骤5.4:将e
kl
送到一个softmax激活函数,通过a
kl
=exp(e
kl
)/∑
l
exp(e
kl
)等式得到归一化的注意力系数a
kl
。
40.步骤5.5:利用得到的a
kl
,通过x
″′
p,k
=∑
lakl
x
″
p,l
等式将每个高阶氨基酸特征更新为相邻高阶氨基酸特征的线性组合,并且随后将relu激活函数添加到x
″′
p,k
。
41.步骤5.6:应用门增强算法,通过前一个高阶氨基酸特征x
′
p,k
和x
″′
p,k
的线性组合使用方程来获得基于门增强的注意力层的输出高阶氨基酸特征zk表示为zk=σ((x
′
p,k
||x
″′
p,k
)u)并解释为x
′
p,k
和x
″′
p,k
传递给的权重系数,其中u是可学习向量,σ表示sigmoid激活函数,(
·
||
·
)是拼接两个向量的操作符。
42.步骤5.7:通过等式,获得最终输出蛋白质特征的向量
43.进一步地,步骤6.1:将输出的配体特征向量和输出的蛋白质特征向量连接起来以表示靶标-配体复合物特征。
44.步骤6.2:随后将特征送到含logsoftmax激活函数的全连接层和含logsoftmax激活函数的输出层。
45.步骤6.3:在输出层之前还添加了一个dropout层,以克服训练过程中的过拟合问题。在训练过程之外执行额外的softmax运算从而归一化模型输出。
46.本发明与现有技术相比,主要有以下有益效果:
47.(1)首先,文献中的大多数研究都考虑了ic
50
性质。然而,这些ic
50
值是噪声数据,随实验条件变化很大,不同研究组之间并不统一。嘈杂的数据不利于训练深度学习模型。因此,我们的工作中去除了ic
50
性质,以使我们的深度学习模型更加合理。
48.(2)其次,选择基于文本的配体和蛋白质分子表示。文本表示方法不高度依赖于靶标-配体复合物的三维结构,与三维分子表示相比,文本表示方法在生成模型输入时可节省更多计算时间成本。针对配体,没有选择常用的基于文本的smiles字符串,因为一个配体可能存在多个不同的smiles字符串进行表示,这可能会导致表示配体时存在一些不确定性。为了避免这个问题,我们将smiles字符串转换为基于文本的mol2vec描述符来表示配体,因为mol2vec由一组独特的morgen片段组成,每一片段均嵌入了特征向量,与基于smiles的文本表示方法相比具有更高的可解释性。此外,用高阶氨基酸字符串表示蛋白质,与常用的氨基酸序列表示相比,高阶氨基酸表示方法可描述氨基酸之间的相互作用,因此更加合理。
49.(3)第三,采用基于门增强的注意力机制和卷积神经网络这两种最先进的技术来有效地提取配体和蛋白质的特征,以加速我们的深度学习模型的学习过程,并在一定程度上减少了计算量。此外,门增强算法可以提高模型的预测精度。
50.基于这三个指标,我们的深度学习模型具有良好的分类能力,可以快速准确地识别具有高结合亲和力的靶标-配体复合物。该方法能够用于辅助药物设计和虚拟筛选,可以大量减少与实验分析相关的时间和成本,提高药物设计和虚拟筛选的效率。
附图说明
51.图1为本发明的流程图;
52.图2为本发明的训练过程、混淆矩阵和受试者工作特征(roc)曲线,其中,(a)为深度学习模型训练集和验证集随轮(epochs)变化的交叉熵损失(cel),(b)为深度学习模型训练集和验证集随轮(epochs)变化的roc曲线下面积(auc),(c)为测试集的二元分类混淆矩阵,(d)为测试集的roc曲线。
具体实施方式
53.以下结合附图和实施例对本发明的流程以及效果进行解释说明。本发明的实施例是在以本发明技术方案为前提下进行实施的,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述实施例。
54.参阅图1,本发明实施例具体公开一种端到端的基于门控注意力机制的靶标-配体复合物结合亲和力预测方法,包括以下步骤:
55.步骤1:根据以下标准对pdbbind数据库(2019版,http://www.pdbbind.org.cn/index.php)进行了处理,建立了一个结合亲和力数据库,用于后续的深度学习模型:
56.删除涉及ic
50
性质的条目,因为这些ic
50
值是噪声数据,随实验条件变化很大,不同研究组之间不统一。嘈杂的数据不利于训练深度学习模型。请注意,ki和kd可以近似地视为相同的性质,并且不随实验条件变化而变化。因此,涉及ki和kd性质的条目将保留在结合亲和力数据库中。删除rdkit工具读取配体结构文件失败或mol2vec方法找不到配体morgen片段的条目。分别删除morgen片段大于140或氨基酸数大于1000的靶标-配体复合物条目。阈值取值判断原则是通过经验来判断的;请注意,阈值的使用能够减少配体和蛋白质矩阵
的最大行数,从而提高深度学习模型的训练效率,代价是消除具有过多morgen片段或氨基酸的少数靶标-配体复合物。k
i/d
《1μm(μmol/l)的靶标-配体复合物标记为高结合亲和力(用“1”表示),而k
i/d
≥1μm的靶标-配体复合物标记为低结合亲和力(用“0”表示),k
i/d
表示ki或kd。最后,在应用上述标准后,总共获得了8,393个独特的靶标-配体复合物。获得的样本存储在我们的结合亲和力数据库中,用于后续的深度学习模型。
57.步骤2:将配体的smiles字符串转换为配体矩阵,作为深度学习模型的输入(图1中的(a)):
58.先将配体的smiles字符串转换为mol2vec描述符,mol2vec描述符学习分子子结构的向量表示。它首先使用morgan算法(半径=1)将分子的smiles字符串表示转换为morgen片段(类似于基团的分子子结构)。然后,将mol2vec中的morgen片段的特征向量嵌入到步骤2.1得到的morgen片段中。将每个分子的morgen片段的特征向量求和来表示整个分子的特征向量。基于mol2vec描述符,每个配体样本由一个140
×
300的二维矩阵表示,其中矩阵行表示morgen片段,矩阵列表示片段的特征向量。考虑到并非每个配体都有140个morgen片段,因此使用填充策略将“0”值添加到没有morgen片段的矩阵行中。请注意,mol2vec中的morgen片段比基于文本的smiles字符串更具可解释性。
59.步骤3:将蛋白质的氨基酸序列转换为蛋白质矩阵,作为深度学习模型的输入(图1中的(b)):
60.通常,蛋白质可以由氨基酸字符串组成的有序序列表示,将蛋白质序列中每三个相邻的氨基酸视为一个高阶氨基酸。例如,如果蛋白质序列是“met-gly-arg-pro-leu”,那么高阶氨基酸是“met-gly-arg”、“gly-arg-pro”、“arg-pro-leu”。通过这种方式,我们的结合亲和力数据库中的蛋白质总共生成了9,164个高阶氨基酸字符串。然后,通过深度学习模型中的嵌入方法,给每个高阶氨基酸字符串嵌入一个特征向量(特征大小设置为30)。最后,每个蛋白质样本由一个1,000
×
30的二维矩阵表示,其中矩阵行表示高阶氨基酸字符串,矩阵列表示高阶氨基酸字符串的特征向量。考虑到并非每个蛋白质都有1,000个高阶氨基酸,因此还使用填充策略将“0”值添加到没有高阶氨基酸的矩阵行中。
61.步骤4:有了获得的亲和力标签(参见步骤1)和输入表示(参见步骤2和3),下一步是构建用于模型训练的深度学习模型的架构。一方面,将配体矩阵送到一个全连接(fc)层和一个基于门增强的注意力层用于特征提取(图1中的(c)),通过矩阵行的加和方式来聚合配体片段的特征向量:
62.首先将配体矩阵x
l
(140
×
300)送到fc层v(300
×
30)以将配体特征的维数从300降低到30,这有助于减少模型冗余并节省训练成本。然后将获得的配体矩阵x
′
l
(140
×
30)送到基于门增强的注意力层。在我们的注意力层中,x
′
l
首先通过一个可学习的权重矩阵w1(30
×
30)进行变换,以获得具有显著特征的新配体矩阵x
″
l
。然后通过方程得到注意力系数e
ij
,其中e
ij
表示第j个片段特征对第i个片段特征的重要性,通过求和和来强制e
ij
等于e
ji
。之后,e
ij
被送到一个softmax激活函数,通过a
ij
=exp(e
ij
)/∑jexp(e
ij
)等式得到归一化的注意力系数a
ij
。利用得到的a
ij
,通过x
″′
l,i
=∑
jaij
x
″
l,j
等式将每个片段特征更新为相邻片段特征的线性组合,并且随后将relu激活函数添加到x
″′
l,i
。
接下来,应用门增强算法,通过前一个片段特征x
′
l,i
和x
″′
l,i
的线性组合使用方程来获得基于门增强的注意力层的输出片段特征zi表示为zi=σ((x
′
l,i
||x
″′
l,i
)u)并解释为x
′
l,i
和x
″′
l,i
传递给的权重系数,其中u是可学习向量(60
×
1),σ表示sigmoid激活函数,(
·
||
·
)是拼接两个向量的操作符。最后,通过等式,将配体片段的特征向量相加成一个表示配体特征的向量。
63.步骤5:另一方面,将蛋白质矩阵先送到一维卷积conv1层和最大池化maxpool层,以减少蛋白质矩阵的行数,然后再送入基于门增强的注意力层用于特征提取,通过这种方式节省注意层不必要的计算时间成本(图1中的(c)),再通过矩阵行的加和方式来聚合蛋白质高阶氨基酸的特征向量:
64.首先将蛋白质矩阵x
p
(1,000
×
30)送到conv1层(输入通道=30,输出通道=30,内核大小=3,步幅=3)和maxpool层(内核大小=3,步幅=3),其中内核用于使用参数共享机制扫描蛋白质矩阵的局部特征。通过这种方式,卷积层和maxpool层能够从整个蛋白质序列中提取关键的高阶氨基酸,这有助于将氨基酸特征集中在靶标-配体复合物的口袋区域,同时减少模型冗余并节省下一个注意力层的计算时间成本。将处理的蛋白质矩阵x
′
p
发送到基于门增强的注意力层。在注意力层中,x
′
p
首先通过一个可学习的权重矩阵w2进行变换,以获得具有显著特征的新蛋白质矩阵x
″
p
。通过方程得到注意力系数e
kl
,其中e
kl
表示第l个高阶氨基酸特征对第k个高阶氨基酸特征的重要性,通过求和和来强制e
kl
等于e
lk
。将e
kl
送到一个softmax激活函数,通过a
kl
=exp(e
kl
)/∑
l
exp(e
kl
)等式得到归一化的注意力系数a
kl
。利用得到的a
kl
,通过x
″′
p,k
=∑
lakl
x
″
p,l
等式将每个高阶氨基酸特征更新为相邻高阶氨基酸特征的线性组合,并且随后将relu激活函数添加到x
″′
p,k
。应用门增强算法,通过前一个高阶氨基酸特征x
′
p,k
和x
″′
p,k
的线性组合使用方程来获得基于门增强的注意力层的输出高阶氨基酸特征zk表示为zk=σ((x
′
p,k
||x
″′
p,k
)u)并解释为x
′
p,k
和x
″′
p,k
传递给的权重系数,其中u是可学习向量,σ表示sigmoid激活函数,(
·
||
·
)是拼接两个向量的操作符。通过等式,获得最终输出蛋白质特征的向量
65.步骤6:将聚合后的配体片段的特征向量与蛋白质高阶氨基酸的特征向量拼接在一起送入后续的fc层以预测靶标-配体复合物的高/低结合亲和力的概率(图1中的(c)):
66.将输出的配体特征向量和输出的蛋白质特征向量拼接起来以表示靶标-配体复合物特征。随后将特征送到含logsoftmax激活函数的fc层(60
×
30)和含logsoftmax激活函数的输出层(fc层)(30
×
2)。基于经验试错发现logsoftmax激活函数可以提升模型的分类效果。在输出层之前还添加了一个dropout层,以克服训练过程中的过拟合问题。请注意,logsoftmax激活函数使输出层的预测总和不等于1。因此,为了预测高/低结合亲和力的概率,在训练过程之外执行额外的softmax运算从而归一化模型输出。
67.实施例
68.靶标-配体复合物以8:1:1的比例随机分为训练集、验证集和测试集。训练集、验证集和测试集的样本数分别为6,711、841和841。训练集中有3,102个低结合亲和力复合物(标签“0”)和3,609个高结合亲和力复合物(标签“1”),满足深度学习分类模型的数据平衡要求。我们的深度学习模型中的损失函数是交叉熵损失(cel),这是分类任务中常用的损失函数。为了最小化损失函数,模型参数通过使用自适应矩估计(adam)优化器进行优化,学习率设置从0.0005到0.00005变化,其中,当验证集的损失函数不随迭代减少时,使用衰减因子(0.5)来更新学习率。一般来说,50%是二分类模型中判断真假的阈值。例如,如果靶标-配体复合物的高结合亲和力的概率为70%,则认为该复合物的结合亲和力是高的(真)。受试者工作特征(roc)曲线会选择一组阈值,并评估每个阈值中的真阳性率和假阳性率。roc曲线下面积(auc)被认为是评估深度学习模型分类标签能力的重要监督函数。auc值相当于随机选择的真样本排名高于随机选择的假样本的概率。因此,它也可以被视为分类模型的不确定性指标。auc越接近1,深度学习模型的分类能力表现越好,越稳定。
69.深度学习模型的超参数根据经验试错确定(如表1所示)。我们使用python语言的开源pytorch库建立深度学习模型。
70.表1深度学习模型的超参数设置
[0071][0072]
我们的深度学习模型的训练结果和性能如表2和图2所示。如表2所示,训练集、验证集、测试集的cel和auc值分别为0.369、0.442、0.401和0.914、0.880、0.901。图2中的(a)和图2中的(b)记录了深度学习模型训练集和验证集随轮(epochs)变化的cel和auc值。上述结果表明,所开发的深度学习模型不存在过拟合问题,并且能够对新样本进行令人满意的泛化预测。请注意,基于门增强的注意力层的引入能够加速我们模型的学习过程。在训练过程中,深度学习模型在第40个轮(epoch)表现良好。如果去除了注意力层,深度学习模型需要更多的轮(epochs)才能获得相似的结果。
[0073]
图2中的(c)是一个基于二元分类的混淆矩阵(2
×
2),用于评估我们的深度学习模型的分类性能,其中x轴上的“0”和“1”代表低结合亲和力和高结合亲和力的标签值,y轴上的“0”和“1”定义了低结合亲和力和高结合亲和力的预测值。混淆矩阵有四种情况,包括真阴性(tn,x=0,y=0),假阴性(fn,x=1,y=0),真阳性(tp,x=1,y=1)和假阳性(fp,x=0,
y=1)。测试样本根据它们的标签值和每个案例的预测值进行计数,图2中的(c)显示tn和tp中的样本数远大于fn和fp中的样本数,这表明开发的深度学习模型能够为区分高/低结合亲和力,即可提供良好的分类能力。此外,图2中的(d)给出了测试集的roc曲线(实线),测试集的auc结果(0.901)也支持我们的深度学习模型具有良好的分类性能。
[0074]
表2深度学习模型的损失函数和度量函数
[0075][0076]
值得注意的是,与其它预测结合亲和力的深度学习模型(例如,kdeep中的参数约1,000,000个)相比,我们所开发的深度学习模型简洁且训练参数(15,634)少很多,表明模型在训练和外推方面的效率。由于以下两个原因,我们的深度学习模型没有与其它深度学习模型进行比较:(1)其它模型中的训练样本涉及ic
50
性质,由于ic
50
是噪声数据并且严重依赖于实验条件,因此在本工作中没有考虑这一性质;(2)我们的深度学习模型本质上是一个分类模型,而其它深度学习模型几乎都是回归模型,用于定量预测结合亲和力大小,性能勉强令人满意(它们的pearson系数在0.80左右)。
[0077]
前述对本发明的具体示例性实施方案的描述是为了说明和例证的目的。这些描述并非想将本发明限定为所公开的精确形式,并且很显然,根据上述教导,可以进行很多改变和变化。对示例性实施例进行选择和描述的目的在于解释本发明的特定原理及其实际应用,从而使得本领域的技术人员能够实现并利用本发明的各种不同的示例性实施方案以及各种不同的选择和改变。本发明的范围意在由权利要求书及其等同形式所限定。以上是对本发明的较佳实施进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明精神的前提下还可做作出种种的等同变形或替换,这些等同的变形或替换均包含在本技术权利要求所限定的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。