1.本技术涉及负极材料技术领域,具体地讲,涉及一种复合负极材料及其制备方法、锂离子电池。
背景技术:
2.锂离子电池由于具备能量密度大、输出功率高、循环寿命长和环境污染小等优点而被广泛应用于电动汽车以及消费类电子产品中。为了提高电池能量密度,高容量的负极材料如si、ge、sn、sb及b等负极材料的研究和开发日趋成熟。然而这些负极材料与锂合金化过程体积膨胀较大,在充放电过程中会粉化从集流体上掉落,使得活性物质与集流体之间失掉电触摸,导致电化学性能变差,容量衰减、循环稳定性下降,难以得到商业应用。
技术实现要素:
3.鉴于此,本技术提出一种能降低膨胀、提高循环稳定性的复合负极材料及其制备方法、锂离子电池。
4.一种复合负极材料,所述复合负极材料包括内核及形成于所述内核表面的包覆层;其中,
5.所述内核为一次颗粒,所述一次颗粒包括骨架,所述骨架包括位于所述一次颗粒的内部的主骨架及自所述主骨架延伸至所述一次颗粒表面的多个分枝。
6.本实施方式的负极材料为一次颗粒,一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝为一个整体,整个骨架结构增强材料的电子传导和离子扩散,可以有效释放锂化后的应力,避免应力在晶界处集中导致材料破裂和粉化。
7.且相较于纳米颗粒堆积而成的二次多孔结构,该负极材料具有一体化结构更加稳定的优点,并可以兼具更小的比表面积和更高的孔隙率。本发明是一次完整颗粒,整个骨架相连,增强材料的电子传到和离子扩散,可以有效释放锂化后的应力,避免应力集中导致材料粉化,二次颗粒组装的多孔材料,由于颗粒之间存在明显的晶界,导致锂化应力集中,颗粒破碎,整个结构破坏,最终导致电化学性能恶化。
8.此外,在内核材料表面的包覆层能够进一步提高负极材料的结构稳定性和循环稳定性,同时进一步缓解负极材料的体积膨胀。
9.一实施方式中,所述主骨架为三维网状结构;
10.一实施方式中,单个所述分枝为单独的晶粒;
11.一实施方式中,所述晶粒的尺寸为30nm-100nm;
12.一实施方式中,所述分枝的横截面最大宽度为20nm-250nm,所述分枝的横截面最大长度为100nm-1500nm;
13.一实施方式中,所述分枝选自棒状纳米颗粒、纳米片、纳米线及纳米管中的至少一种。
14.一种复合负极材料,所述复合负极材料包括内核及形成于所述内核表面的包覆
层;其中,所述内核为一次颗粒,所述一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,所述孔道延伸至所述一次颗粒的表面。
15.该负极材料的孔道延伸至一次颗粒的表面的孔道结构具有如下优点:一、在提高储锂性能的同时能降低了锂电池的膨胀。不仅可以缓解嵌锂过程中的体积膨胀,还有利于为锂化提供内膨胀的空间,使电极材料锂化后向内膨胀而降低整个电极膜的厚度,大大提高了锂离子电池的安全性。二、提供了电解液流动的通道,有利于电解液的接触。孔道结构还可以带来更高的振实密度,能够增加电池的体积能量密度。
16.一实施方式中,所述孔道的直径为10nm-150nm;所述孔道的深度为50nm-1500nm。
17.一种复合负极材料,所述复合负极材料包括内核及形成于所述内核表面的包覆层;其中,所述内核为一次颗粒,所述一次颗粒内部形成有通孔,所述一次颗粒的孔隙率不低于30%。
18.本技术的一次颗粒具有较高的孔隙率,可以有效缓解负极材料300%以上的体积膨胀,结合孔贯通结构优势,锂化后的孔结构也可以保持完好,现有技术的孔隙率较低,无法满足负极材料(如硅)的巨大体积膨胀,锂化后孔洞被填充后,由于电化学烧结最终导致孔洞被填堵,无法继续保持多孔结构。
19.一实施方式中,所述包覆层包括碳层、金属氧化物层及金属氮化物层中的至少一种;
20.所述包覆层包括碳层,以所述复合负极材料质量百分含量为100%计,所述碳的质量百分比含量为2%~50%;
21.一实施方式中,所述包覆层包括石墨烯层及非晶碳层中的至少一种;
22.一实施方式中,所述包覆层包括石墨烯层,所述石墨烯层具有褶皱结构;
23.一实施方式中,所述褶皱结构的粗糙度最大峰-谷高度差大于10nm且小于1μm,所述褶皱结构的相邻两波峰或相邻两波谷之间的距离大于10nm且小于1μm;
24.一实施方式中,根据所述褶皱结构的褶皱面弯曲形态分类,所述褶皱结构选自圆弧褶皱、尖棱褶皱、扇状褶皱中的至少一种;
25.一实施方式中,根据所述褶皱结构的轴面产状和两翼产状分类,所述褶皱结构选自直立褶皱、斜歪褶皱、倒转褶皱和平卧褶皱中的至少一种;
26.一实施方式中,所述包覆层包括非晶碳层,所述非晶碳层的厚度为5nm~150nm;
27.一实施方式中,所述包覆层包括金属氧化物层,以所述复合负极材料质量百分含量为100%计,所述金属氧化物的质量百分比含量为2%~60%;
28.一实施方式中,所述包覆层包括金属氧化物层,所述金属氧化物层中的金属元素包括ti、v、nb、ta、w和zr中的至少一种;
29.一实施方式中,所述金属氧化物层中的金属元素与氧元素的摩尔比为1:(0.1-3);所述金属氧化物层的厚度为1nm-200nm;
30.一实施方式中,所述包覆层包括金属氮化物层,以所述复合负极材料质量百分含量为100%计,金属氮化物的质量百分比含量为2%~70%。
31.一实施方式中,所述包覆层包括金属氮化物层,所述金属氮化物层中的金属元素包括ti、v、nb、ta、w和zr中的至少一种,
32.一实施方式中,所述金属氮化物层厚度为1nm~250nm。
33.一实施方式中,所述一次颗粒选自硅、锗、锑、锡、硼中的至少一种;
34.一实施方式中,所述一次颗粒的中值粒径为0.2μm~15μm;
35.一实施方式中,所述一次颗粒的比表面积为5m2/g~100m2/g;
36.一实施方式中,所述一次颗粒的孔隙率为30%~70%;
37.一实施方式中,所述一次颗粒的粉体振实密度为0.2g/cm3~0.8g/cm3;
38.一实施方式中,所述一次颗粒的粉体压实密度为1.2g/cm3~1.8g/cm3;
39.一实施方式中,所述复合负极材料的中值粒径为0.1μm~15μm;
40.一实施方式中,所述复合负极材料的比表面积为1m2/g~150m2/g;
41.一实施方式中,所述复合负极材料的孔隙率为30%~70%。
42.一种复合负极材料的制备方法,包括以下步骤:
43.将含有n-m合金和含卤素的六元环有机物的混合物置于保护性气氛中进行置换反应,得到反应产物,所述反应产物包括m的氧化物、m的卤化物;
44.去除所述m的氧化物及m的卤化物,得到复合负极材料;
45.所述n-m合金中的n包括硅、锗、锑、锡、硼中的至少一种;
46.所述n-m合金中的m包括镁、铝、钙和锌中的至少一种。
47.该制备方法通过一步复合法制备负极材料,高温下n-m合金与六元环有机物直接反应,去除n-m合金中的金属m成分,同时在n材料表面原位沉积碳层。整体反应温和且没有副产物,n材料结构完整、稳定,碳层沉积均匀,参与反应的原材料都是常用的合金、有机物以及金属盐,能够降低成本。
48.一实施方式中,所述n-m合金和所述含卤素的六元环有机物的摩尔比为1:(0.2~6);
49.一实施方式中,所述含卤素的六元环有机物包括卤代环己烷及其衍生物、卤代苯、卤代苯甲酸和卤代苯胺中的至少一种,且所述卤素包括氟、氯和溴中的至少一种;
50.一实施方式中,当所述含卤素的六元环有机物使用卤代环己烷时,所述混合物还包括裂解抑制剂,其中,所述裂解抑制剂包括酰胺类化合物和氰酸盐;
51.一实施方式中,所述硅合金与所述酰胺类化合物的摩尔比为1:(0.1-10);所述硅合金与所述氰酸盐的摩尔比为1:(0.1-10);
52.一实施方式中,所述酰胺类化合物包括碳酰胺、甲酰胺、乙酰胺、二甲基甲酰胺及内酰胺中的至少一种;
53.所述氰酸盐包括氰酸钾、氰酸钠和氰酸铵中的至少一种;
54.一实施方式中,所述置换反应的反应温度为200℃~1000℃,反应时间1h~24h;
55.一实施方式中,所述保护性气氛包括氦气、氖气、氩气、氪气及氙气中的至少一种;
56.一实施方式中,所述去除所述m的氧化物及m的卤化物的方法包括酸洗,所述酸洗采用的酸溶液的质量浓度为1mol/l~5mol/l;
57.一实施方式中,所述酸洗采用的酸溶液包括盐酸、硝酸及硫酸中的至少一种。
58.一种复合负极材料的制备方法,包括以下步骤:
59.将复合物在真空环境下进行置换反应,得到反应产物,所述反应产物包括m1的氧化物,所述复合物包括表面形成金属氧化物层的n1-m1材料;
60.去除所述m1的氧化物,得到复合负极材料;
61.所述n1-m1材料中的n1包括硅、锗、锑、锡及硼中的至少一种;
62.所述n1-m1材料中的m1包括镁、铝、钙及锌中的至少一种。
63.在本实施方式中,通过在n1-m1材料表面包覆一层金属氧化层,高温下n1-m1材料与部分金属氧化层发生置换反应,去除n1-m1材料中的m1成分,同时在n1材料表面生成未完全反应的金属氧化层,酸洗后便可得到复合负极材料。在本方案中,金属氧化物层具有刚性好,致密性优等优点,现价与柔性的碳层材料,可以有效抑制n1的体积膨胀而导致的整个结构破坏,减少材料的体积膨胀同时避免电解液与硅负极接触,减少副反应,提高整个复合材料的首效。
64.一种复合负极材料的制备方法,包括以下步骤:
65.将复合物在保护气氛下进行热处理后再进行氮化处理,得到反应产物,所述反应产物包括m1的氧化物,所述复合物包括表面形成金属氧化物层的n1-m1材料;
66.去除所述m1的氧化物,得到复合负极材料;
67.所述n1-m1材料中的n1包括硅、锗、锑、锡及硼中的至少一种;
68.所述n1-m1材料中的m1包括镁、铝、钙及锌中的至少一种。
69.通过在n1-m1材料表面包覆一层金属氧化层后,在保护气氛下热处理,一方面保证n1-m1材料与部分金属氧化层发生置换反应,去除n1-m1材料中的金属m1成分;另一方面,保证氧化物在高温常压下由非晶转化为晶体。进一步通过氮化处理使得在内核表面的金属氧化层被氮化为金属氮化物。在本方案中,金属氮化物层不仅刚性好,而且导电性优异,可以有效缓解硅的体积膨胀同时增加材料的导电性,提高材料的倍率性,减少材料不可逆容量损失,带来高的容量。
70.一实施方式中,所述n1-m1材料为金属间化合物及合金中的至少一种;
71.一实施方式中,所述在n1-m1材料表面形成金属氧化物层包括水热法,溶胶凝胶法、沉淀法、化学气相沉积法、磁控溅射及固相反应法中的至少一种;
72.一实施方式中,所述金属氧化物层中的金属元素包括ti、v、nb、ta、w和zr中的至少一种;
73.一实施方式中,所述真空环境下的真空度小于1000pa;
74.一实施方式中,所述还原反应的温度为500℃-1100℃,保温时间1h~48h;
75.一实施方式中,所述热处理温度为500℃-800℃,保温时间1h~24h;所述保护性气氛包括氦气、氖气、氩气、氪气及氙气中的至少一种;
76.一实施方式中,所述氮化处理在400℃-950℃下保温2h~24h;氮化处理的气氛采用氨气气氛、氮气气氛中的至少一种;
77.一实施方式中,所述去除所述m的氧化物的方法为酸洗。
78.一种锂离子电池,所述锂离子电池包括所述的复合负极材料或所述复合负极材料的制备方法制备的负极材料。
附图说明
79.图1为本实施例提供的复合负极材料的结构示意图;
80.图2为本实施例提供的复合负极材料的石墨烯层结构示意图;
81.图3为本实施例提供的复合负极材料的制备方法的流程示意图;
82.图4为本实施例提供的硅碳复合负极材料的合成流程示意图;
83.图5为本实施例提供的硅/金属氧化物及硅/金属氮化物复合负极材料的合成流程示意图;
84.图6a为本实施例1提供的硅碳复合负极材料的扫描电镜图片;
85.图6b为本实施例1提供的硅碳复合负极材料的另一扫描电镜图片;
86.图7为本实施例1提供的硅碳复合负极材料的xrd图;
87.图8为本实施例1提供的硅碳复合负极材料的拉曼图;
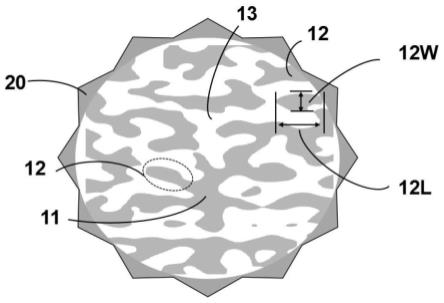
88.图9本实施例1提供的硅碳复合负极材料的循环性能曲线图。
具体实施方式
89.以下所述是本发明实施例的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明实施例原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明实施例的保护范围。
90.目前,在锂离子电池中,负极材料是影响其充放电性能的关键材料之一,现有的负极材料为石墨类碳材料,碳材料的理论储锂容量仅为372ma h/g,无法满足人们对高能量密度材料的需求。负极材料如si、ge、sn、sb及b等由于具有高的质量容量和较低的电压平台,可以带来高的能量密度。然而这些负极材料与锂合金化过程中会产生巨大的体积膨胀,导致负极材料粉化,失去电接触,容量衰减迅速,如硅作为锂离子电池负极材料具有很高的理论容量(约4200ma h/g),但是硅负极材料脱嵌锂过程中体积膨胀较大(》300%),在充放电过程中易粉化而从集流体上掉落,使得活性物质与集流体之间失掉电触摸,导致电化学性能变差。因此,大的体积变化效应容易导致其具有较差的循环稳定性,难以商业化应用。为了提高锂离子电池的循环稳定性,本技术实施例提供了一种膨胀低、稳定性好的复合负极材料。
91.具体地,如图1所示,复合负极材料包括内核10及形成于内核10表面的包覆层20;其中,
92.内核10为一次颗粒,一次颗粒包括骨架,骨架包括位于一次颗粒的内部的主骨架11及自主骨架11延伸至所述一次颗粒表面的多个分枝12。
93.请进一步参阅图1,该实施方式的内核10为一次颗粒,一次颗粒为大孔结构,一次颗粒的内部形成有孔道13,孔道13延伸至一次颗粒的表面。其中,根据国际纯粹与应用化学协会(iupac)的定义,孔径大于50nm的称为大孔。
94.本实施方式的负极材料为一次颗粒,一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝为一个整体,整个骨架结构增强材料的电子传导和离子扩散,可以有效释放锂化后的应力,避免应力在晶界处集中导致材料破裂和粉化。
95.该负极材料的孔道延伸至一次颗粒的表面的孔道结构具有如下优点:一、在提高储锂性能的同时能降低了锂电池的膨胀。不仅可以缓解嵌锂过程中的体积膨胀,还有利于为锂化提供内膨胀的空间,使电极材料锂化后向内膨胀而降低整个电极膜的厚度,大大提高了锂离子电池的安全性。二、提供了电解液流动的通道,有利于电解液的接触。孔道结构还可以带来更高的振实密度,能够增加电池的体积能量密度。
96.且相较于纳米颗粒堆积而成的二次多孔结构,本实施方式所制备的负极材料具有
一体化结构更加稳定的优点,并可以兼具更小的比表面积和更高的孔隙率。本发明是一次完整颗粒,整个骨架相连,增强材料的电子传到和离子扩散,可以有效释放锂化后的应力,避免应力集中导致材料粉化,二次颗粒组装的多孔材料,由于颗粒之间存在明显的晶界,导致锂化应力集中,颗粒破碎,整个结构破坏,最终导致电化学性能恶化。
97.此外,在内核材料表面的包覆层能够进一步提高负极材料的结构稳定性和循环稳定性,同时进一步缓解负极材料的体积膨胀。
98.在一些实施方式中,主骨架为三维网状结构;
99.在一些实施方式中,单个所述分枝为单独的晶粒;分支与主骨架之间没有明显的晶界,一次颗粒的表面分散晶粒,现有技术方案中多孔硅二次颗粒上的分枝主要由多个小晶粒组成,晶界较多,相较于这种结构,本实施方式中一次颗粒上的分枝为单独一个大晶粒,没有过多的晶界存在,锂化后应力可以得到较好的分散,避免应力集中导致材料的破坏;同时单独一个晶粒的晶面曲线相同,更有利于减小材料某一方向的相对体积膨胀,而多个小晶粒组成的结构体积膨胀相对较大,同时结构稳定性也相对较差,导致循环稳定性较差。
100.具体地,晶粒的尺寸为30nm-100nm;示例性地,晶粒的尺寸可以是30nm、45nm、50nm、60nm、75nm、100nm。
101.请继续参阅图1,分枝12的横截面的最大宽度12w为20nm~250nm,分枝12的横截面的最大长度12l为100nm~1500nm。
102.示例性地,分枝12的横截面的最大宽度12w例如可以是20nm、40nm、80nm、100nm、120nm、150nm、180nm、200nm或250nm,分枝12的横截面的最大长度12l例如可以是100nm、200nm、300nm、400nm、500nm、800nm、1000nm、1200nm或1500nm,在此不做限定。
103.在一些实施方式中,分枝选自棒状纳米颗粒、纳米片、纳米线及纳米管中的至少一种。
104.在一些实施方式中,通过压汞测试方法测得孔道13的直径为10nm-150nm;孔道的深度为50nm-1500nm。示例性地,孔道直径具体可以为10nm、50nm、60nm、80nm、100nm或150nm,在此不做限定。孔道13深度具体可以是50nm、100nm、200nm、300nm、400nm、500nm、800nm或1000nm,在此不做限定。可以控制反应时间与反应温度来改变孔的深度(一般反应时间越长、反应温度越高,孔的深度越深。)
105.在一些实施方式中,一次颗粒选自硅、锗、锡、硼及锑中的至少一种;其骨架可以为硅骨架、锗骨架、锡骨架、硼骨架、锑骨架等;如一次颗粒选自硅材料,一次颗粒包括硅骨架,包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。锗、硼、锡及锑的骨架结构与上述硅骨架结构相似。
106.在一些实施方式中,一次颗粒的中值粒径为0.2μm~15μm,例如0.2μm、1μm、3μm、5μm、8μm、10μm、12μm或15μm等。优选为0.5μm~10μm,进一步优选为1μm~5μm。
107.在一些实施方式中,一次颗粒的比表面积为5m2/g~00m2/g,例如5m2/g、10m2/g、20m2/g、30m2/g、40m2/g、50m2/g、60m2/g、80m2/g或100m2/g等。优选为10m2/g~50m2/g。
108.在一些实施方式中,一次颗粒的粉体振实密度为0.2g/cm3~0.8g/cm3,例如0.2g/cm3、0.3g/cm3、0.5g/cm3、0.6g/cm3、0.7g/cm3或0.8g/cm3等。优选为0.4g/cm3~0.7g/cm3。
109.在一些实施方式中,一次颗粒的粉体压实密度为1.2g/cm3~1.8g/cm3,例如1.2g/
cm3、1.3g/cm3、1.4g/cm3、1.5g/cm3、1.6g/cm3或1.8g/cm3等,优选为1.4g/cm3~1.7g/cm3;
110.在一些实施方式中,复合负极材料的中值粒径为0.1μm~15μm,可选地,复合负极材料的中值粒径具体可以是0.1μm、0.5μm、1μm、2μm、3μm、4μm、5μm、6μm、7μm、8μm、9μm或10μm等,在此不做限定。复合负极材料的中值粒径优选为0.5μm~10μm,更优选为1μm~8μm。
111.复合负极材料的比表面积比为1m2/g~150m2/g。可选地,复合负极材料的比表面积比可以是1m2/g、5m2/g、10m2/g、20m2/g、30m2/g、40m2/g、50m2/g、60m2/g、70m2/g、100m2/g、120m2/g或150m2/g等,在此不做限定;复合负极材料的比表面积比优选为1m2/g~50m2/g。可以理解地,比表面积越小越好,过大的比表面积容易导致sei膜形成,消耗不可逆锂盐过多,降低电池的首次效率低,综合考虑制备工艺的成本,将比表面积控制在10m2/g~50m2/g。
112.在一些实施方式中,包覆层包括碳层、金属氧化物层及金属氮化物层中的至少一种;碳层以及金属氧化物层和氮化物层,能够进一步提高负极材料的导电性、结构稳定性和循环稳定性,同时进一步缓解负极材料的体积膨胀;
113.其中,包覆层包括碳层,进一步地,碳层包括石墨烯层及非晶碳层中的至少一种;
114.更进一步地,石墨烯层具有褶皱结构,石墨烯层的褶皱形貌能够提供更多的活性位点,从而进一步提升负极材料的导电性、倍率性能;
115.如图2所示,褶皱结构包括凸起(峰)201和凹陷(谷)202,相邻两个凸起201之间为凹陷202,相邻两个凹陷202之间为凸起201;其中,该褶皱结构的表面粗糙度rz(最大峰-谷高度)大于10nm且小于1μm,进一步地,rz可大于50nm且小于500nm,更进一步地,rz可大于100nm且小于350nm。相近两凸起最高点或两凹陷最低点之间的距离h大于10nm且小于1μm;进一步地,h大于50nm且小于800μm,更进一步地,h大于100nm且小于500nm。
116.具体地,褶皱结构根据褶皱面弯曲形态分类,褶皱结构可选自圆弧褶皱、尖棱褶皱及扇状褶皱中的至少一种;更进一步地,褶皱结构可同时具有圆弧褶皱、尖棱褶皱及扇状褶皱。
117.褶皱结构根据轴面产状和两翼产状分类,褶皱结构可选自直立褶皱、斜歪褶皱、倒转褶皱及平卧褶皱中的至少一种;更进一步地,褶皱结构可同时具有直立褶皱、斜歪褶皱、倒转褶皱和平卧褶皱。
118.石墨烯层具有上述形态的褶皱结构,能够提供更多的活性位点,从而进一步提升负极材料的导电性、倍率性能。
119.在一些实施方式中,碳层包括非晶碳层,非晶碳层的厚度为5nm~150nm;
120.在一些实施方式中,以所述复合负极材料质量百分含量为100%计,碳的质量百分比含量为2%~50%;
121.在一些实施方式中,包覆层包括金属氧化物层,金属氧化物层中的金属包括ti、v、nb、ta、w和zr中的至少一种;
122.其中,金属氧化物层中的金属元素与氧元素的摩尔比为1:(0.1-3);金属氧化物层的厚度为1nm-200nm;
123.以所述复合负极材料质量百分含量为100%计,金属氧化物的质量百分比含量为2%~60%;
124.在一些实施方式中,包覆层包括金属氮化物层,金属氮化物层中的金属元素包括ti、v、nb、ta、w和zr中的至少一种,进一步地,金属氮化物层厚度为1nm~250nm。
125.以复合负极材料质量百分含量为100%计,金属氮化物的质量百分比含量为2%~70%。
126.金属氧化物层及其氮化物层充当刚性保护壳,避免一次颗粒的体积膨胀撑破整个材料,保证优异的结构稳定性和长的循环寿命。
127.另一方面,复合负极材料包括内核10及形成于内核10表面的包覆层20;内核10为一次颗粒,一次颗粒内部形成有通孔,一次颗粒的孔隙率不低于30%。本发明的一次颗粒具有较高的孔隙率,可以有效缓解负极材料300%以上的体积膨胀,结合大孔贯通结构优势,锂化后的孔结构也可以保持完好,现有技术的孔隙率较低以及存在较多的微孔和介孔,无法满足负极材料(如硅)的巨大体积膨胀,锂化后孔洞被填充后,由于电化学烧结最终导致孔洞被填堵,无法继续保持多孔结构。
128.进一步地,一次颗粒的孔隙率为30%~70%,例如30%、35%、40%、50%、55%、60%或70%等,优选为40%~60%。
129.该实施方式的一次颗粒与包覆层如前所述,在此不做重复描述。
130.当包覆层为碳层时,一实施方式的一种复合负极材料的制备方法,如图4所示,所述方法包括以下步骤s100~s400:
131.s100、制备n-m合金;
132.在一些实施方式中,n-m合金中的n包括si、ge、sn、b及sb中的至少一种;n-m合金中的m包括mg、al、zn和ca中的至少一种。具体的示例中,n-m合金可以为si-mg合金、si-al合金、ge-mg合金、ge-al合金等。不同种类的合金可得到不同形状的分枝,包括棒状纳米颗粒、纳米片、纳米线及纳米管中的至少一种。
133.在一些实施方式中,n-m合金制备方法为将n粉和活泼金属m混合后在保护性气体下加热反应,制得n-m合金。
134.其中,所述n粉的粉末粒径为0.1μm~15μm,具体可以是0.1μm、0.5μm、1μm、3μm、5μm、8μm、10μm或15μm等,在此不做限定。
135.活泼金属m的粉末粒径为0.1μm~80μm,具体可以是0.1μm、5μm、10μm、20μm、40μm、50μm或80μm等,在此不做限定。
136.n粉与活泼金属m的摩尔比为1:(1~3),具体可以是1:1、1:1.5、1:2、1:2.5或1:3,在此不作限定。
137.加热反应的温度为400℃~900℃,例如可以是400℃、500℃、600℃、700℃、800℃或900℃。
138.加热反应的保温时间为2h~8h,例如可以是2h、4h、6h或8h,在此不做限定。
139.加热反应的升温速率为1℃/min~10℃/min,例如可以是1℃/min、3℃/min、5℃/min、8℃/min或10℃/min,在此不作限定。
140.在本技术中,通过控制硅粉粒径、活泼金属粒径、反应温度、反应时间等参数,有利于n-m合金的生成,提高n-m合金的金属元素的掺杂均匀性。
141.当然,n-m合金还可以通过其他制备方法制备,例如:高能球磨、真空冶炼以及热压烧结等。可以理解,n-m合金可以通过市售获得,此时步骤s100可以省略。
142.在一些实施例中,n-m合金中的n的质量百分比含量为15%~60%,可选地,n的质量百分比含量可以是15%、20%、30%、40%、50%或60%,在此不做限定。在具体实施例中,
n-m合金例如可以是硅镁合金、硅铝合金、硅钙合金、硅锌合金中的至少一种。可以理解地,可以通过控制n-m合金的成分来改变n材料的孔道的孔的大小以及孔隙率,一般n-m合金中n含量越高,孔径越小。可以控制加热反应时间与反应温度来改变孔的深度,一般反应时间越长、反应温度越高,孔的深度越深。
143.在具体实施例中,所述方法还包括:
144.将制得的所述n-m合金进行粉碎处理,调整n-m合金粉末的粒径尺寸至0.1μm~15μm,例如可以是0.1μm、0.5μm、1μm、2μm、5μm、10μm或15μm,在此不做限定。
145.具体地,所述粉碎处理的设备包括行星式球磨机、砂磨机及气流粉粹机中的至少一种。可以理解地,n-m合金粒径越小,其比表面积更大,去合金化热处理时能够反应能够更充分。
146.s200、将n-m合金和含卤素的六元环有机物进行混合,得到混合物。
147.具体地,所述n-m合金和所述含卤素的六元环有机物的摩尔比为1:(0.2-6),例如可以是1:0.2、1:0.5、1:1、1:2、1:3、1:4、1:5或1:6,在此不做限定。
148.具体地,所述含卤素的六元环有机物包括卤代环己烷及其衍生物、卤代苯、卤代苯甲酸和卤代苯胺中的至少一种。所述卤素包括氟、氯和溴中的至少一种。
149.卤代环己烷为六元环状烃,其结构稳定性相较于卤代苯更差,在高温下容易发生烃链断裂。卤代环己烷例如可以是氯代环己烷、六氯环己烷、六溴环己烷、三氯环己烷。卤代苯例如可以是三溴苯、六氯苯、六溴苯等等。卤代苯甲酸例如可以是氯苯甲酸、溴苯甲酸、2-溴苯甲酸、4-溴苯甲酸、3-氯苯甲酸等等。卤代苯胺例如可以是对氯苯胺、4-溴苯胺、2-氯苯胺等等。
150.当含卤素的六元环有机物采用卤代环己烷时,进行步骤s300之前,所述方法还包括步骤s201:
151.往所述混合物中加入裂解抑制剂,其中,所述裂解抑制剂包括酰胺类化合物和氰酸盐。所述酰胺类化合物包括碳酰胺、甲酰胺、乙酰胺、二甲基甲酰胺及内酰胺中的至少一种;所述氰酸盐包括氰酸钾、氰酸钠及氰酸铵中的至少一种。
152.需要说明的是,碳酰胺加热至(150℃~160℃)就会分解脱氨成为异氰酸。氰酸钾在高温(700℃~900℃)且隔绝空气时能够分解为氰氨化钾与二氧化碳,同样地,氰酸钠在550℃左右分解为氰氨化钠与二氧化碳。可以理解地,分解出来的二氧化碳气体、无机盐对环境没有危害,裂解抑制剂在高温且隔绝空气时能够发生分解反应,吸收部分反应能量,从而抑制卤代环己烷或其衍生物发生自身裂解。
153.在具体实施例中,所述n-m合金与所述酰胺类化合物的摩尔比为1:(0.1-10),例如可以是1:0.1、1:0.5、1:1、1:5或1:10等,在此不做限定。所述硅合金与所述氰酸盐的摩尔比为1:(0.1-10),例如可以是1:0.1、1:0.5、1:1、1:5或1:10等,在此不做限定。
154.s300、将所述混合物在保护性气氛下进行置换反应,得到反应产物,反应产物包括m的氧化物、m的卤化物;
155.在具体实施例中,为了混合物充分反应,置换反应的温度为200℃~1000℃,例如可以是200℃、300℃、400℃、600℃、800℃或950℃。
156.保温时间1h~24h,例如可以是1h、3h、6h、9h、12h、15h、18h或24h,在此不做限定。
157.其中,当温度为200~750℃时,生成的碳层为非晶碳层;当大于750℃时,生成的碳
层为褶皱石墨烯。
158.置换反应的加热速率为1℃/min~20℃/min,例如可以是1℃/min、5℃/min、10℃/min、15℃/min或20℃/min。从而有效提高反应效率。
159.可以理解地,在上述适宜的热处理的温度、时间、升温速率范围内,有助于提高去合金效率,有助于硅合金在去合金过程中形成骨架结构。
160.为了提高反应的安全性,在保护性气氛条件下进行置换反应,所述保护性气氛的气体包括氮气、氦气、氖气、氩气和疝气中的至少一种。保护性气体的流量可以控制在1l/min-10l/min。
161.s400、去除m的氧化物、m的卤化物,得到复合负极材料。
162.需要说明的是,反应产物包括m的卤化物、m的氧化物、碳及n,因此,需要去除反应产物中的金属氧化物及卤化物。
163.作为本技术可选的技术方案,去除方法包括酸洗,将反应产物进行酸洗,可以去除反应产物中的金属氧化物及卤化物。
164.可选地,所述酸洗采用的酸溶液包括盐酸、硝酸及硫酸中的至少一种。
165.所述酸溶液的质量浓度为1mol/l~5mol/l,例如可以是1mol/l、2mol/l、3mol/l、4mol/l或5mol/l。当然也可以根据实际需求调制酸溶液的质量浓度,在此不做限定。
166.所述酸洗的时长为1h~10h,例如可以是1h、3h、5h、7h或10h。在本实施例中,酸洗的产物仍可以回收循环使用。
167.在本方案中,通过一步复合法制备负极材料,高温下n-m合金与六元环有机物直接反应,去除n-m合金中的金属m成分,同时在n材料表面原位沉积碳层。整体反应温和且没有副产物,n材料结构完整、稳定,碳层沉积均匀。参与反应的原材料都是常用的合金、有机物以及金属盐,能够降低成本。
168.在本方案中,石墨烯层和非晶碳层是原位生长形成的。以含有卤素的六元环有机物为碳源和n-m合金在一步去合金制备出复合负极材料,该方法利用n-m合金中在高温下产生的金属蒸气具有较强的还原性,与六元环有机物发生反应后去除n-m合金中的金属m成分,自身六元环原位生长且有序自组装形成褶皱石墨烯层或者非晶碳层,酸洗后得到褶皱石墨烯或者非晶碳层包裹n材料,采用一步复合法制备得到复合负极材料,能够有效提高制备效率,制备方法简单易行,绿色环保,制备过程可控程度高,成本低且可以大规模生产。制备得到复合负极材料具有容量高、充放电循环稳定性高、膨胀率低等优点,能够广泛应用于锂电池中。需要说明的是,一步复合法是指硅材料制备与碳包覆同时进行,其制备效率高于两步复合法。
169.该实施方式的方法制备得到的复合负极材料包括内核10及形成于内核10表面的包覆层20;其中,
170.内核10为一次颗粒,一次颗粒包括骨架,骨架包括位于一次颗粒的内部的主骨架11及自主骨架11延伸至所述一次颗粒表面的多个分枝12。
171.该实施方式的内核10为一次颗粒,一次颗粒为大孔结构,一次颗粒的内部形成有孔道13,孔道13延伸至一次颗粒的表面;
172.包覆层20为碳包覆层。
173.示例性地,图5为硅碳复合负极材料的合成流程示意图。
174.当包覆层为金属氧化物层时,一实施方式的一种复合负极材料的制备方法,所述方法包括以下步骤s100
′
~s400
′
:
175.s100
′
、制备n1-m1材料;
176.在一些实施方式中,n1-m1材料为金属间化合物及合金中的至少一种;
177.其中,n1-m1材料中的n1包括硅、锗、锑、锡、硼中的至少一种;n1-m1材料中的m1包括镁、铝、钙和锌中的至少一种。
178.具体地,n1-m1合金的制备方法同步骤s100;在此不做详细描述;
179.s200
′
、在n1-m1材料表面形成金属氧化物层,得到复合物;
180.在具体实施例中,金属氧化物层的金属元素包括ti、v、nb、ta、w和zr中的至少一种。金属氧化物层中金属元素与氧元素的摩尔比为1:(0.1-3);
181.在一些实施方式中,金属氧化物层的厚度为1nm-200nm。
182.具体地,所述n1-m1材料和所述金属氧化物层的金属氧化物的摩尔比为1:(0.01-5),例如可以是1:0.02、1:0.05、1:1、1:2、1:3、1:4或1:5,在此不做限定。
183.具体地,在n1-m1材料表面形成金属氧化物层方法采用本领域常规的方法,如可为水热法,溶胶凝胶法、沉淀法、化学气相沉积法、磁控溅射或固相反应法;
184.s300
′
、将复合物至于真空环境下进行置换反应,得到反应产物;
185.具体地,反应产物包括m1的氧化物、n1、金属氧化物;
186.在一些实施方式中,真空环境下的真空度小于1000pa;
187.置换反应的温度为500℃~1100℃,例如可以500℃、600℃、700℃、800℃、900℃或1200℃。
188.置换反应的保温时间为1h~48h,例如可以是1h、24h、36h或48h,在此不做限定。
189.还原反应的升温速率为1℃/min~20℃/min,例如可以是1℃/min、3℃/min、5℃/min、8℃/min或20℃/min,在此不作限定。
190.s400
′
、去除所述m1的氧化物,得到复合负极材料;
191.具体地,去除m1的氧化物的方法包括酸洗;
192.进一步地,酸洗采用的酸溶液包括盐酸、硝酸及硫酸中的至少一种。
193.酸溶液的质量浓度为1mol/l~5mol/l,例如可以是1mol/l、2mol/l、3mol/l、4mol/l或5mol/l。当然也可以根据实际需求调制酸溶液的质量浓度,在此不做限定。
194.酸洗的时长为1h~10h,例如可以是1h、3h、5h、7h或10h。在本实施例中,酸洗的产物仍可以回收循环使用。
195.在本实施方式中,通过在n1-m1材料表面包覆一层金属氧化层,高温下n1-m1材料与部分金属氧化层发生置换反应,去除n1-m1材料中的m1成分,同时在n1材料表面生成未完全反应的金属氧化层,酸洗后便可得到复合负极材料。在本方案中,金属氧化物层具有刚性好,致密性优等优点,现价与柔性的碳层材料,可以有效抑制n1的体积膨胀而导致的整个结构破坏,减少材料的体积膨胀同时避免电解液与硅负极接触,减少副反应,提高整个复合材料的首效。
196.本发明实施例提供的复合负极材料的制备方法制备得到复合负极材料能够有效提高锂电池充放电循环的稳定性,具有高容量、长循环寿命、首效高以及低膨胀等多个优点。
197.该实施方式的方法制备得到的复合负极材料包括内核10及形成于内核10表面的包覆层20;其中,
198.内核10为一次颗粒,一次颗粒包括骨架,骨架包括位于一次颗粒的内部的主骨架11及自主骨架11延伸至所述一次颗粒表面的多个分枝12。
199.该实施方式的内核10为一次颗粒,一次颗粒为大孔结构,一次颗粒的内部形成有孔道13,孔道13延伸至一次颗粒的表面;
200.包覆层20为金属氧化层。
201.当包覆层为金属氮化物层时,一实施方式的一种复合负极材料的制备方法,所述方法包括以下步骤s100
″
~s400
″
:
202.s100
″
、制备n1-m1材料;
203.s200
″
、在n1-m1材料表面形成金属氧化物层,得到复合物;
204.其中步骤s100
″
与s200
″
如同上述步骤s100
′
与s200
′
,在此不做重复描述。
205.s300
″
、将复合物在保护气氛下进行热处理后再进行氮化处理,得到反应产物;
206.其中,反应产物包括m1的氧化物、n1、金属氧化物;
207.其中,保护气氛中热处理温度为500-800℃,保温时间1h~24h,热处理的反应加热速率为1~20℃/min;
208.保护性气氛包括氦气、氖气、氩气、氪气及氙气中的至少一种;
209.具体地,氮化处理中,处理温度为400-950℃,保温时间为2h~24h;氮化气氛为氨气气氛、等离子氮气气氛中的至少一种。
210.s400
″
、去除m1的氧化物,得到复合负极材料;
211.去除m1的氧化物的方法如同步骤s400
′
在此不做重复描述。
212.该实施方式的方法制备得到的复合负极材料包括内核10及形成于内核10表面的包覆层20;其中,
213.内核10为一次颗粒,一次颗粒包括骨架,骨架包括位于一次颗粒的内部的主骨架11及自主骨架11延伸至所述一次颗粒表面的多个分枝12。
214.该实施方式的内核10为一次颗粒,一次颗粒为大孔结构,一次颗粒的内部形成有孔道13,孔道13延伸至一次颗粒的表面;
215.包覆层20为金属氮化层。
216.在本实施方式中,通过在n1-m1材料表面包覆一层金属氧化层后,在保护气氛下热处理,一方面保证n1-m1材料与部分金属氧化层发生置换反应,去除n1-m1材料中的金属m1成分;另一方面,保证氧化物在高温常压下由非晶转化为晶体。进一步通过氮化处理使得在内核表面的金属氧化层被氮化为金属氮化物,紧接着酸洗后便可得到复合负极材料。在本方案中,金属氮化物层不仅刚性好,而且导电性优异,可以有效缓解硅的体积膨胀同时增加材料的导电性,提高材料的倍率性,减少材料不可逆容量损失,带来高的容量。
217.示例性地,图5为硅/金属氧化物及硅/金属氮化物复合负极材料的合成流程示意图;
218.本发明实施例提供的复合负极材料的制备方法制备得到复合负极材料能够有效提高锂电池充放电循环的稳定性,具有高容量、长循环寿命、倍率性能好以及低膨胀等多个优点。
219.本发明实施例还提供了一种锂离子电池负极极片和一种锂离子电池,采用本发明上述实施例提供的复合负极材料或采用本发明上述实施例提供的复合负极材料的制备方法制得的负极材料。
220.下面分多个实施例对本发明实施例进行进一步的说明。其中,本发明实施例不限定于以下的具体实施例。在不变主权利的范围内,可以适当的进行变更实施。
221.实施例1
222.一种硅碳复合负极材料的制备方法,包括以下步骤:
223.(1)将粒径为1μm的硅粉与镁粉按摩尔比为1:2混合均匀后放入气氛炉,在氩气惰性气体保护下以3℃/min的加热速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到1μm的硅镁合金粉末。
224.(2)将1mol的硅镁合金粉末与1mol的六氯环己烷混合均匀,再加入碳酰胺、氰酸钾和氰酸钠各1mol,得到混合物。
225.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到10pa,然后将反应釜在氩气气氛中以3℃/min的加热速率加热到780℃后保温8h使其充分反应,得到反应产物;
226.(4)将反应产物在1mol/l的盐酸溶液机械搅拌酸洗处理2h后,抽滤、洗涤、干燥后得到硅碳复合负极材料。
227.所得的硅碳复合负极材料的中值粒径约为1μm,比表面积为53m2/g,孔隙率为48%,碳的质量百分比含量为12%。
228.图6a、图6b为硅碳复合负极材料的扫描电镜图片,图7为本实施例提供的硅碳复合负极材料的拉曼图;图8为本实施例提供的硅碳复合负极材料的xrd图,从图6a、图6b中的扫描电镜图片可以看出,所得产物的硅碳复合负极材料包括内核及形成于所述内核表面的包覆层;其中,
229.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至所述一次颗粒的表面。
230.包覆层为2nm褶皱石墨烯。孔道的平均直径约为100nm,孔道的深度约为700nm。
231.图7的拉曼图谱进一步证明在2680cm-1
附近存在一个2d峰,这是石墨的特征峰,进一步说明原位生成的碳为类石墨碳,具有更好的导电性和稳定性。从图8的xrd图谱中可以看出在28.4
°
、47.3
°
和56.1
°
的三强峰与硅(jcpds no.27-1402)的三强峰相对应,基本无杂相。
232.实施例2
233.一种硅碳复合负极材料的制备方法,包括以下步骤:
234.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到650℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
235.(2)将1mol的硅镁合金粉末与1mol的六氯苯混合均匀,得到混合物。
236.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到650℃后保温8h使其充分反应,得到反应
产物;
237.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅复合负极材料。
238.硅碳复合负极材料的中值粒径约为0.6μm,比表面积为79m2/g,孔隙率为56%,碳的质量百分比含量为20%。
239.硅碳复合负极材料包括内核及形成于内核表面的包覆层;其中,
240.内核为一次颗粒,一次颗粒包括硅骨架,骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至所述一次颗粒的表面。
241.包覆层为10nm非晶碳层,孔道的平均直径约为130nm,孔道的深度约为800nm。
242.实施例3
243.一种硅碳复合负极材料的制备方法,包括以下步骤:
244.(1)将12μm的硅粉与镁粉按摩尔比为1:2混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到10μm的硅镁合金粉末;
245.(2)将1mol的硅镁合金粉末与1mol的六氯苯混合均匀,得到混合物。
246.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到800℃后保温8h使其充分反应,得到反应产物;
247.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅复合负极材料。
248.所得的硅碳复合负极材料的中值粒径约为10μm,比表面积为43m2/g,孔隙率为61%,碳的质量百分比含量为15%;
249.硅碳复合负极材料包括内核及形成于内核表面的包覆层;其中,
250.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面。
251.包覆层为5nm褶皱石墨烯层,三维孔道的平均直径约为110nm,三维孔道的孔深度约为670nm。
252.实施例4
253.一种硅碳复合负极材料的制备方法,包括以下步骤:
254.(1)将1μm的硅粉与镁粉按摩尔比为1:1.2混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到1μm的硅镁合金粉末;
255.(2)将1mol的硅镁合金粉末与0.5mol的六氯苯混合均匀,得到混合物。
256.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到10pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到800℃后保温8h使其充分反应,得到反应产物;
257.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗
涤、干燥后得到复合负极材料。
258.所得的硅碳复合负极材料的中值粒径约为1μm,比表面积为38m2/g,孔隙率为47%,碳的质量百分比含量为5%。
259.硅碳复合负极材料包括内核及形成于内核表面的包覆层;其中,
260.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为4nm褶皱石墨烯层;孔道的平均直径约为65nm,三维孔道的深度约为680nm。
261.实施例5
262.一种硅碳复合负极材料的制备方法,包括以下步骤:
263.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到800℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
264.(2)将1mol的硅镁合金粉末与4mol的六氯苯混合均匀,得到混合物。
265.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到750℃后保温8h使其充分反应,得到反应产物;
266.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到复合负极材料。
267.所得的硅碳复合负极材料的中值粒径约为0.5μm,比表面积为80m2/g,孔隙率为53%,碳的质量百分比含量为22%。
268.硅碳复合负极材料包括内核及形成于内核表面的包覆层;其中,
269.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为6nm褶皱石墨烯层。孔道的平均直径约为130nm,孔道的深度约为940nm。
270.实施例6
271.一种锗碳复合负极材料的制备方法,包括以下步骤:
272.(1)将1.5μm的锗粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到480℃后保温6h,使其充分反应得到锗镁合金;将锗镁合金球磨后得到0.5μm的锗镁合金粉末;
273.(2)将1mol的锗镁合金粉末与3mol的六氯苯混合均匀,得到混合物。
274.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到600℃后保温8h使其充分反应,得到反应产物;
275.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到复合负极材料。
276.所得的锗碳复合负极材料的中值粒径约为0.6μm,比表面积为75m2/g,孔隙率为63%,碳的质量百分比含量为18%。
277.锗碳复合负极材料包括内核及形成于内核表面的包覆层;其中,
278.内核为一次颗粒,一次颗粒包括锗骨架,锗骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为12nm非晶碳层。三维孔道的平均孔直径约为120nm,三维孔道的孔深度约为35nm。
279.实施例7
280.一种硅碳复合负极材料的制备方法,包括以下步骤:
281.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
282.(2)将1mol的硅镁合金粉末、2.6mol的氯代环己烷及0.5mol的碳酰胺混合均匀,得到混合物。
283.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到750℃后保温8h使其充分反应,得到反应产物;
284.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅复合负极材料。
285.所得的硅碳复合负极材料的中值粒径约为0.5μm,比表面积为64m2/g,孔隙率为53%,碳的质量百分比含量为19%。
286.硅碳复合负极材料包括内核及形成于内核表面的包覆层;其中,
287.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为3.2nm褶皱石墨烯层,孔道的平均直径约为110nm,孔道的深度约为780nm。
288.实施例8
289.一种硅复合负极材料的制备方法,包括以下步骤:
290.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
291.(2)将1mol的硅镁合金粉末、0.7g纤维素溶于500ml无水乙醇中分散均匀后得到混合溶液。
292.(3)将3g钛酸四丁酯合逐滴加入到上述混合溶液中,加热至80℃后快速搅拌3小时后进行抽滤,在60℃条件下真空干燥24小时后得到氧化钛前驱体包裹硅镁合金复合物;
293.(4)将上述复合物放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到750℃后保温8h使其充分反应,得到反应产物;
294.(4)将反应产物放入在4l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅/氧化钛复合负极材料。
295.所得的复合负极材料的中值粒径约为0.6μm,比表面积为44m2/g,孔隙率为43%,氧化钛的质量百分比含量为12%。
296.复合负极材料包括内核及形成于内核表面的包覆层;其中,
297.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为氧化钛层。孔道的平均直径约为20nm,孔道的深度约为150nm。
298.实施例9
299.一种硅复合负极材料的制备方法,包括以下步骤:
300.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
301.(2)将1mol的硅镁合金粉末、0.7g纤维素溶于500ml无水乙醇中分散均匀后得到混合溶液。
302.(3)将4g钛酸四丁酯合逐滴加入到上述混合溶液中,加热至80℃后快速搅拌3小时后进行抽滤,在60℃条件下真空干燥24小时后得到氧化钛前驱体包裹硅镁合金复合物;
303.(4)将上述复合物放入管式气氛炉中,在氩气气氛中以3℃/min的升温速率加热到650℃后保温5h使其充分反应,接着将氩气换成氨气后将温度升到800℃后保温8h后,得到反应产物;
304.(4)将反应产物放入在4l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅/氮化钛复合负极材料。
305.所得的复合负极材料的中值粒径约为0.7μm,比表面积为40m2/g,孔隙率为48%,氮化钛的质量百分比含量为32%。
306.复合负极材料包括内核及形成于内核表面的包覆层;其中,
307.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为氮化钛层。孔道的平均直径约为50nm,孔道的深度约为400nm。
308.实施例10
309.一种硅复合负极材料的制备方法,包括以下步骤:
310.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
311.(2)通过溶胶-凝胶过程制备五氧化二钒包裹硅镁合金,首先配置0.15m/l浓度的三异丙氧基氧化钒的酒精溶液500ml,加入30ml的乙酰丙酮,得到混合溶液;将0.5mol的硅镁合金粉末加入上述混合溶液中搅拌36小时后分散均匀后得到溶胶。
312.(3)溶胶进行抽滤,在60℃条件下真空干燥24小时后得到五氧化二钒前驱体包裹硅镁合金复合物;
313.(4)将上述复合物放入管式气氛炉中,在氩气气氛中以3℃/min的升温速率加热到600℃后保温3h使其充分反应,接着将氩气换成氨气后将温度升到750℃后保温8h后,得到反应产物;
314.(5)将反应产物放入在4l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅/氮化钒复合负极材料。
315.所得的复合负极材料的中值粒径约为0.6μm,比表面积为51m2/g,孔隙率为40%,氮化钛的质量百分比含量为25%。
316.复合负极材料包括内核及形成于内核表面的包覆层;其中,
317.内核为一次颗粒,一次颗粒包括硅骨架,硅骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为60nm氮化钒层。孔道的平均直径约为125nm,孔道的深度约为350nm。
318.实施例11
319.一种复合负极材料的制备方法,包括以下步骤:
320.(1)将1.5μm的锗粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到锗镁合金;将锗镁合金球磨后得到0.5μm的锗镁合金粉末;
321.(2)将1mol的锗镁合金粉末、0.7g纤维素溶于500ml无水乙醇中分散均匀后得到混合溶液。
322.(3)将3g钛酸四丁酯合逐滴加入到上述混合溶液中,加热至80℃后快速搅拌3小时后进行抽滤,在60℃条件下真空干燥24小时后得到氧化钛前驱体包裹锗镁合金复合物;
323.(4)将上述复合物放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到750℃后保温8h使其充分反应,得到反应产物;
324.(4)将反应产物放入在4l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔锗/氧化钛复合负极材料。
325.所得的复合负极材料的中值粒径约为0.55μm,比表面积为41m2/g,孔隙率为45%,氧化钛的质量百分比含量为13%。
326.复合负极材料包括内核及形成于内核表面的包覆层;其中,
327.内核为一次颗粒,一次颗粒包括锗骨架,锗骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为氧化钛层。孔道的平均直径约为600nm,孔道的深度约为180nm。
328.实施例12
329.一种复合负极材料的制备方法,包括以下步骤:
330.(1)将1.5μm的锗粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到600℃后保温6h,使其充分反应得到锗镁合金;将锗镁合金球磨后得到1.0μm的锗镁合金粉末;
331.(2)通过溶胶-凝胶过程制备五氧化二钒包裹锗镁合金,首先配置0.1m/l浓度的三异丙氧基氧化钒的酒精溶液500ml,加入30ml的乙酰丙酮,得到混合溶液;将0.5mol的锗镁合金粉末加入上述混合溶液中搅拌36小时后分散均匀后得到溶胶。
332.(3)溶胶进行抽滤,在60℃条件下真空干燥24小时后得到五氧化二钒前驱体包裹锗镁合金复合物;
333.(4)将上述复合物放入管式气氛炉中,在氩气气氛中以3℃/min的升温速率加热到600℃后保温3h使其充分反应,接着将氩气换成氨气后将温度升到750℃后保温8h后,得到反应产物;
334.(5)将反应产物放入在4l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔锗/氮化钒复合负极材料。
335.所得的复合负极材料的中值粒径约为1.2μm,比表面积为52m2/g,孔隙率为43%,氮化钛的质量百分比含量为24%。
336.复合负极材料包括内核及形成于内核表面的包覆层;其中,
337.内核为一次颗粒,一次颗粒包括锗骨架,锗骨架包括位于一次颗粒的内部的主骨架及自主骨架延伸至所述一次颗粒表面的多个分枝。一次颗粒为大孔结构,所述一次颗粒的内部形成有孔道,孔道延伸至一次颗粒的表面;包覆层为50nm氮化钒层。孔道的平均直径约为150nm,孔道的深度约为900nm。
338.实施例13
339.一种复合负极材料的制备方法,包括以下步骤:
340.(1)将1.5μm的硅粉与镁粉按摩尔比为1:2.5混合均匀后放入气氛炉,在氩气惰性气体保护下以5℃/min的升温速率加热到650℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到0.5μm的硅镁合金粉末;
341.(2)将1mol的硅镁合金粉末与0.1mol的六氯苯混合均匀,得到混合物。
342.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到100pa,然后将反应釜在氩气气氛中以3℃/min的升温速率加热到750℃后保温8h使其充分反应,得到反应产物;
343.(4)将反应产物放入在2l的1mol/l盐酸溶液中机械搅拌酸洗处理3h后,抽滤、洗涤、干燥后得到多孔硅复合负极材料。
344.所得的硅碳复合负极材料的中值粒径约为0.6μm,比表面积为79m2/g,孔隙率为56%,碳的质量百分比含量为20%,孔道的平均直径约为130nm,孔道的深度约为800nm。
345.实施例14
346.(1)将粒径为1μm的硅粉与镁粉按摩尔比为1:2混合均匀后放入气氛炉,在氩气惰性气体保护下以3℃/min的加热速率加热到600℃后保温6h,使其充分反应得到硅镁合金;将硅镁合金球磨后得到1μm的硅镁合金粉末。
347.(2)将1mol的硅镁合金粉末与2.6mol的六氯环己烷混合均匀,得到混合物。
348.(3)将所得混合物混合均匀后放入密封不锈钢反应釜中,抽真空到10pa,然后将反应釜在氩气气氛中以3℃/min的加热速率加热到750℃后保温8h使其充分反应,得到反应产物;
349.(4)将反应产物在1mol/l的盐酸溶液机械搅拌酸洗处理2h后,抽滤、洗涤、干燥后得到硅碳复合负极材料。
350.所得的硅碳复合负极材料的中值粒径约为1μm,比表面积为77m2/g,孔隙率为56%,碳的质量百分比含量为19%,一次粒子的中值粒径约为0.5μm,三维孔道的平均孔直径约为100nm,三维孔道的孔深度约为730nm,不能形成石墨烯层,碳层厚度25nm。
351.对比例1
352.采用硅碳复合负极材料sio/c,硅碳复合负极材料的中值粒径为1.5μm,复合材料中内核为纳米硅粒子堆积的多孔硅结构,外壳为碳层包裹;其中,碳的质量百分比含量为22%,比表面积为38m2/g,碳层厚度为50nm,硅碳复合负极材料的孔隙率为67%。即为siox/c负极材料,其中x=1.0。
353.综上所述,实施例1~14与对比例1制得的负极材料,样本编号对应为s1~s14及r1;负极材料的性能参数如表1所述:
354.表1
[0355][0356]
性能测试:
[0357]
将负极材料分别与羧甲基纤维素钠、丁苯橡胶以及导电石墨(ks-6)和碳黑(sp)按照比例92:2:2:2:2配置浆料,均匀涂覆与铜箔上烘干制成负极极片,在氩气气氛手套箱中组装成扣式电池,所用隔膜为聚丙烯微孔膜,所用电解液为1mol/l的六氟磷酸锂(溶剂为碳酸乙烯酯、碳酸甲乙酯和碳酸二甲酯的混合液),所用对电极为金属锂片。
[0358]
对上述15组电池在蓝电ct2001a电池测试系统上进行放电比容量测试,1小时放电的电量与电池容量的比为放电比容量。
[0359]
对上述15组电池在蓝电ct2001a电池测试系统上进行首次库伦效率测试,充放电电流为0.05c,测得首次库伦效率。
[0360]
对上述15组电池在蓝电ct2001a电池测试系统上进行循环100周测试,充放电电流为0.2c,循环100圈后测试计算圈后电池容量及圈后容量保持率。
[0361]
其中,0.2c循环100圈后容量保持率=第100圈循环放电容量/第一周放电容量*100%,结果如表2所示。
[0362]
表2各电池的参数性能比对表
[0363][0364]
本技术的复合负极材料具有低膨胀、循环稳定佳优点。
[0365]
由上表1~2可知,实施例13与实施例2的主要区别在于,硅镁合金与六氯苯的混合比例较低,导致硅材料表面的石墨烯层厚度大幅下降,不利于增加负极材料的导电性且对材料的体积膨胀抑制性能较弱,导致长循环性能差。
[0366]
实施例14与实施例1的主要区别在于,没有加入裂解抑制剂(碳酰胺、氰酸钾和氰酸钠),氯代环己烷在反应过程中大量分解,难以在硅材料表面沉积形成石墨烯层,导致电池的充放电比容量、首次库伦效率、圈后容量保持率等各项性能有所下降。对比例1是碳包覆的多孔硅负极材料,该材质制成的电池的0.2c循环100圈后容量及容量保持率均有所下降,并且对比例1的电极膜膨胀率也高于实施例1。
[0367]
综上所述,本技术提供的硅碳复合负极材料的制备方法,简单易操作,制备过程安全、高效;制造成本有效降低,适于量化生产;制得的产物用作电池极片使用,具有较好的充放电循环性能。
[0368]
本技术虽然以较佳实施例公开如上,但并不是用来限定权利要求,任何本领域技术人员在不脱离本技术构思的前提下,都可以做出若干可能的变动和修改,因此本技术的保护范围应当以本技术权利要求所界定的范围为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。