1.本发明属于生物信息学领域,具体涉及一种融合网络拓扑信息的化合物-蛋白质相互作用预测方法。
背景技术:
2.蛋白质是生物体实现生命活动的基础,在生物体中发挥着广泛而重要的作用。药物通常是具备特殊性质的化合物,它们通过与生物体中的特定蛋白质结合而影响蛋白质的功能,进而产生药物功效。化合物-蛋白质相互作用的研究是药物设计的重要组成部分,对药物开发具有重要意义。为了提高药物研发效率,许多基于深度学习的预测模型被开发出来,但现有模型未能将网络拓扑信息显式地融合到模型中。
技术实现要素:
3.本发明的目的是提供一种融合网络拓扑信息的化合物-蛋白质相互作用预测方法,能够有效地提高化合物-蛋白质相互作用预测的准确率。
4.为实现上述目的,本发明采用如下的技术方案:
5.一种融合网络拓扑信息的化合物-蛋白质相互作用预测方法,包括以下步骤:
6.步骤1:对数据进行预处理;
7.步骤2:根据数据集构建化合物-蛋白质相互作用网络,计算相互作用网络中每个节点的度,作为节点的中心性度量;
8.步骤3:对于数据集中每对化合物和蛋白质,计算化合物在相互作用网络中的各相邻节点与蛋白质的共同邻居数,作为化合物对蛋白质的相关性度量;同理得到蛋白质对化合物的相关性度量;
9.步骤4:构建一个基于transformer的二分类模型,根据得到的节点的中心性度量为每个节点分配一个实值嵌入向量,将其添加到节点特征中。
10.步骤5:为得到的每对节点的相关性的每个可能取值分别分配一个可学习标量,将其作为步骤4所述模型中交叉注意模块的偏置项。
11.步骤6:最后利用全连接层,输出预测概率。
12.进一步的,所述步骤1具体为:
13.步骤1.1:对化合物-蛋白质相互作用数据,蛋白质序列信息,化合物smiles数据进行预处理,去掉异常值,随机生成负例,随机划分数据集;
14.步骤1.2:使用seqvec模型编码蛋白质序列;
15.步骤1.3:使用rdkit提取化合物特征和化合物图的邻接矩阵。
16.进一步的,所述步骤2具体为:
17.步骤2.1:将原始数据集中的每个化合物和每个蛋白质都作为节点,成对的化合物和蛋白质的正相互作用作为边,构建化合物-蛋白质相互作用网络。
18.步骤2.2:计算网络中每个节点的邻居节点的数目,作为节点的度中心性。
19.进一步的,所述步骤3具体为:
20.步骤3.1:计算并保存相互作用网络中所有蛋白质两两之间的共同邻居以及所有化合物两两之间的共同邻居数。
21.步骤3.2:对于数据集中每对化合物和蛋白质,根据步骤3.1保存的结果,查找化合物在相互作用网络中的各相邻节点与蛋白质的共同邻居数,记录其中的最大值作为化合物对蛋白质的相关性度量。
22.步骤3.3:对于数据集中每对化合物和蛋白质,根据步骤3.1保存的结果,查找蛋白质在相互作用网络中的各相邻节点与化合物的共同邻居数,记录其中的最大值作为蛋白质对化合物的相关性度量。
23.进一步的,所述步骤4具体为:
24.步骤4.1:构建一个传统的transformer模型,去除解码器的位置编码并将掩码由下三角矩阵变换成邻接矩阵以使解码器只能看到相邻节点。
25.步骤4.2:根据节点的中心性度量为每个节点分配一个实值嵌入向量,将其添加到节点特征中,方法如下:
26.f=x z
deg
ꢀꢀ
(1)
27.其中,f代表最终得到的新的特征向量。x代表氨基酸或者原子的初始特征向量。z是可学习的嵌入向量,由蛋白质或化合物节点的度指定。
28.进一步的,所述步骤5具体为:
[0029][0030]
其中,是传统的注意力权重计算方法,函数φ由节点之间的相关性来定义,是一个可学习标量,由函数φ的输出值索引,并在所有层中共享。
[0031]
本发明与现有技术相比具有以下有益效果:
[0032]
1、本发明考虑到了网络拓扑信息,在模型中融合了节点中心性编码和相关性编码,使模型能够包含更多有效信息;
[0033]
2、构建了基于transformer的二分类模型,利用其中的交叉注意力机制处理蛋白质特征和化合物特征之间的关系,以融合多模态信息,提高了相互作用预测的准确性。
附图说明
[0034]
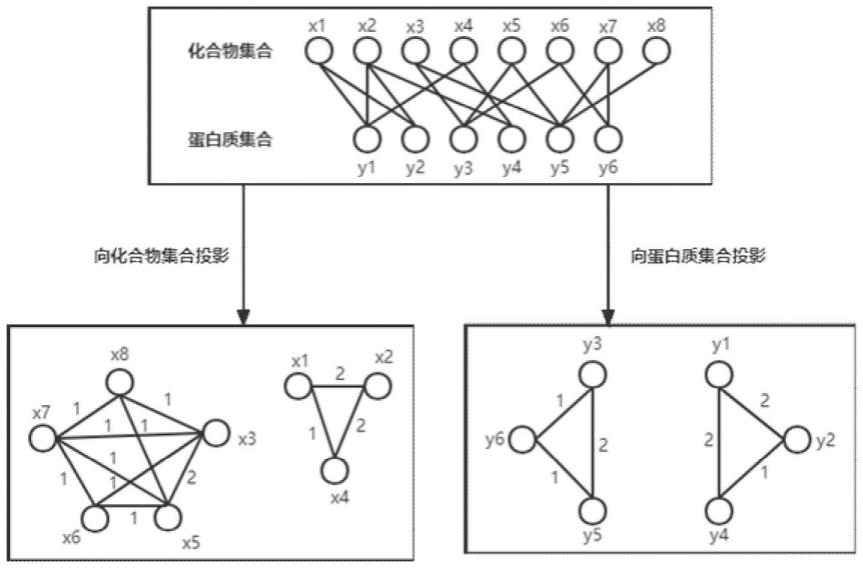
图1是节点相关性计算方法示意图;
[0035]
图2是系统的原理示意图;
[0036]
图3a是在人类数据集上本发明实施例与其他方法性能比较的折线图;
[0037]
图3b是在秀线虫数据集上本发明实施例与其他方法性能比较的折线图。
具体实施方式
[0038]
下面结合附图及实例对本发明做进一步说明。
[0039]
如图1所示,本发明提出了一种融合网络拓扑信息的化合物-蛋白质相互作用预测方法,首先,根据化合物和蛋白质的网络拓扑信息,计算每个节点的中心性和成对数据的相
关性,然后建立模型预测化合物-蛋白质之间潜在的相互作用关系。最后采用相应指标对本模型的性能进行了评估。为了使相关性计算方法能够更加直观地表示,本发明建立了相应的示意图,如图1所示。通过数据集构建化合物-蛋白质相互作用网络,然后对其进行投影操作,通过计算成对数据中节点1在相互作用网络中的各相邻节点与节点2的共同邻居数,得到节点1对节点2的相关性度量。示意图中上半部分表示原始的相互作用网络,可以看出其本质上是一个二部图,下半部分表示原网络向化合物集合的投影和向蛋白质集合的投影,两个蛋白质节点如果与同一个化合物相连,则将两个蛋白质连接起来,边的权重为共同化合物的个数。对于化合物节点也进行相同的处理。根据获得的投影计算相关性,如图1所示,要求x1对y1的相关性,那么首先要找到x1的除去y1之外的其它邻居中,与y1的共同邻居最多的那个节点,取其共同邻居数。在这里,x1只有两个邻居,只有y2符和条件,y2与y1的共同邻居有2个,分别是x1和x2,所以x1对y1的相关性为2。然后再找到y1的其它邻居中,与x1的共同邻居最多的那个节点,是图1中的x2,取x2和x1的共同邻居数2,作为y1对x1的相关性。最后,为求得的每个可能的值分别分配一个可学习标量,它将作为交叉注意模块中的偏置项,即为相关性编码,为模型增加了更多的有效信息。本发明的目的即为利用融合了中心性编码和相关性编码的化合物-蛋白质相互作用预测模型来对潜在的相互作用进行预测。
[0040]
如图2所示,本发明的具体实施例流程为:
[0041]
步骤1:首先从human和caenorhabditis elegans数据集中获取化合物-蛋白质的相互作用对,human数据集包含了1052种化合物和852种蛋白质之间的3369种正相互作用;caenorhabditis elegans数据集包含1434种化合物和2504种蛋白质之间的4000种正相互作用。去掉异常值后,随机生成与正样本数目相同的负样本,并随机划分训练、验证和测试集。通过seqvec模型获得氨基酸嵌入,seqvec是用大型蛋白质数据库对elmo进行预训练得到的。将分子的smiles描述符通过一个用于化学信息学的开源工具包rdkit获得原子嵌入。
[0042]
步骤2:分别将两个数据集中的每个化合物和每个蛋白质都作为节点,成对的化合物和蛋白质的正相互作用作为边,构建化合物-蛋白质相互作用网络,计算网络中每个节点的邻居节点的数目,作为节点的度中心性。
[0043]
步骤3:对于数据集中每对化合物和蛋白质,计算化合物在相互作用网络中的各相邻节点与蛋白质的共同邻居数,作为化合物对蛋白质的相关性度量,代表能与该化合物发生相互作用的蛋白质们与目标蛋白质拥有的相同的化合物个数,直觉上,就是两个已经有很多共同化合物的蛋白质节点,有更强的趋势在未来拥有更多的共同化合物;同理得到蛋白质对化合物的相关性度量。
[0044]
步骤4:构建一个基于transformer的二分类模型,去除解码器的位置编码并将掩码由下三角矩阵变换成邻接矩阵以使解码器只能看到相邻节点。根据得到的节点的中心性度量为每个节点分配一个实值嵌入向量,将其与节点的原始特征矩阵做加法,如公式(1),作为新的特征矩阵输入所述模型。
[0045]
f=x z
deg
ꢀꢀ
(1)
[0046]
步骤5:为得到的每对节点的相关性的每个可能取值分别分配一个可学习标量,将其作为步骤4所述模型中交叉注意模块的偏置项,如公式(2)。
[0047]
[0048]
其中,是传统的注意力权重计算方法,函数φ由节点之间的相关性来定义,是一个可学习标量,由函数φ的输出值索引,并在所有层中共享。
[0049]
步骤6:最后利用全连接层,输出预测概率。
[0050]
本发明的有效性验证:
[0051]
通过对比实验,分别在5个指标上对本发明进行性能评估,本发明与其他方法比较的结果如图3a和图3b所示,其中,本发明在测试集上的最好指标达到了精准率:0.997,召回率:1,准确性:0.999,f1score:0.998,和auc:1。验证结果表明,本发明的性能均优于其他方法。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。