1.本发明涉及一种嘧啶甲酰胺类化合物及其应用。
背景技术:
2.vanin-1(血管非炎性分子-1)是一种具有泛肽酶活性的外切酶,主要催化泛酰巯基乙胺的水解而产生泛酸(pantothenic acid,vb5)和巯基乙胺。由vb5合成而来的辅酶a (coa)调节脂肪酸合成和氧化以及能力代谢等生物转化,而巯基乙胺和胱胺之间的可逆反应是氧化应激的重要传感器。越来越多的研究发现,巯基乙胺的缺乏或水平降低导致γ-gcs活性增强,引起组织中内源性gsh储备升高,从而可以预防或消除组织炎症。研究发现,vanin-1的mrna在人结肠、十二指肠、子宫内膜、肝、肾、胆囊和小肠中高表达。在uc(溃疡性结肠炎)患者中,vanin-1高表达且弥漫,而且仅限于笔刷边界。另外,在uc的临床静止期,结肠中vanin-1的表达水平仍显著高于对照品。在tnbs模型实验中,小鼠vanin-1敲除(vanin-1-/-)后生存率明显高于模型对照组,并且未表现明显体重降低。而且,90%经胱胺处理的vanin-1-/-小鼠在5天内死亡,说明胱胺完全逆转 vanin-1的缺乏对结肠炎的保护作用。此外,对小鼠组织病理学分析发现,对vanin-1的抑制或敲除能显著改善小鼠结肠的病变(berruyer c,等人vanin-1-/-mice exhibit aglutathione mediated tissue resistance to oxidative stress.mol.cell biol.2004;24:7214-7224; berruyer c,等人vanin-1licenses inflammatory mediator production by gut epithelial cells andcontrols colitis by antagonizing peroxisome proliferator-activated receptorγactivity.j.exp.med. 2006;203:2817-2827)。
3.另外,vanin-1也被认为在心血管疾病和肿瘤疾病中起调节作用。有研究证明,vanin
‑ꢀ
1在体外调节平滑肌细胞的活化,并在体内调节响应于颈动脉结扎的新内膜增生的发生。 vnn1基因的多态性与血压和hdl水平相关。在sf-1转基因小鼠中,vanin-1缺失防止小鼠发展为肾上腺皮质的瘤形成,表明了vanin-1在某些癌症中的作用。在炎症性疾病方面的研究发现,与正常个体相比,在牛皮癣的皮肤损伤中,vanin-1高度上调。在患有儿童免疫血小板减少症(itp)的患者的全血中,vnn1的基因表达也上调,其中vnn1的过表达与慢性itp的进展相关。另外,在患有多种肾脏病症的患者尿液中已检测到vanin-1 升高,所述肾脏病症包括系统性红斑狼疮、肾脏毒物(nephrotoxicant)-诱导的肾损伤和2型糖尿病(rommelaere s,等人pparalpha regulates the production of serum vanin-1by liver. febs lett.2013nov 15;587(22):3742-8)。
技术实现要素:
4.本发明所要解决的技术问题是为了克服现有技术中基于vanin酶的治疗剂缺乏的问题;而提供了一种嘧啶甲酰胺类化合物及其应用。本发明提供的嘧啶甲酰胺类化合物为具有vanin酶抑制活性的化合物;具有较强的vanin-1抑制剂活性;其可以用于治疗多种病状,包括克罗恩氏病及溃疡性结肠炎。
5.本发明是通过下述技术方案来解决上述技术问题的。
6.本发明提供了一种如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐;
[0007][0008]
其中,r
a1
、r
a3
和r
a5
独立地为h、卤素、cn、c
1-c6烷基、c
1-c6烷基-o-、被一个或多个r
1a
取代的c
1-c6烷基、或被一个或多个r
1b
取代的c
1-c6烷基-o-;
[0009]ra2
和r
a4
独立地为h、卤素;
[0010]
r4和r5独立地为h、c
1-c6烷基、被一个或多个卤素取代的c
1-c6烷基;或者,r4和 r5与它们键合的碳一起形成3-7元环烷基;
[0011]
r6和r7独立地为h、卤素、c
1-c6烷基、被一个或多个r
1c
取代的c
1-c6烷基、
‑ꢀ
n(r
2ar2b
)或-n(r
3a
)-(c=o)-r
3b
;
[0012]
或者,r6和r7与它们键合的碳一起形成环b;环b为4-7元环烷基、4-7元杂环烷基、被一个或多个r
1d
取代的4-7元环烷基或被一个或多个r
1e
取代的4-7元杂环烷基;所述的4-7元杂环烷基和被一个或多个r
1e
取代的4-7元杂环烷基里的4-7元杂环烷基中,杂原子为n、o或s,杂原子个数为1或2个;
[0013]r1a
、r
1b
、r
1c
、r
1d
和r
1e
独立地为卤素、c
1-c4烷基或被一个或多个卤素取代的c1‑ꢀ
c4烷基;
[0014]r2a
、r
2b
、r
3a
和r
3b
独立地为h或c
1-c4烷基。
[0015]
在本发明某些优选实施方案中,所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐中的某些基团如下定义,未提及的基团同本技术任一方案所述(以下简称为“在某一方案中”),
[0016]ra1
、r
a3
和r
a5
独立地为h、卤素、cn、c
1-c6烷基-o-、被一个或多个r
1a
取代的c1‑ꢀ
c6烷基、或被一个或多个r
1b
取代的c
1-c6烷基-o-;
[0017]
例如h、卤素、c
1-c6烷基-o-、被一个或多个r
1a
取代的c
1-c6烷基、或被一个或多个r
1b
取代的c
1-c6烷基-o-;
[0018]
又例如h、卤素或被一个或多个r
1a
取代的c
1-c6烷基;
[0019]
再例如卤素或被一个或多个r
1a
取代的c
1-c6烷基;
[0020]
还例如卤素。
[0021]
在某一方案中,
[0022]
为
例如
[0023]
在某一方案中,
[0024]r1a
和r
1b
独立地为卤素,例如f或cl,又例如f。
[0025]
在某一方案中,
[0026]r1a
和r
1b
的个数为1、2、3、4或5个,例如2或3。
[0027]
在某一方案中,
[0028]
为
[0029]
r4和r5独立地为c
1-c6烷基;或,r4和r5与它们键合的碳一起形成3-7元环烷基;
[0030]
例如为
[0031]
又例如
[0032]
再例如r4独立地为c
1-c6烷基;或,r4和r5与它们键合的碳一起形成3-7元环烷基。
[0033]
在某一方案中,
[0034]
r6和r7独立地为h、n(r
2ar2b
)或-n(r
3a
)-(c=o)-r
3b
;或,r6和r7与它们键合的碳一起形成环b;
[0035]
例如r6为h,r7为n(r
2ar2b
)或-n(r
3a
)-(c=o)-r
3b
。
[0036]
在某一方案中,
[0037]
r6和r7与它们键合的碳一起形成环b;环b为4-7元杂环烷基。
[0038]
在某一方案中,
[0039]r2a
和r
2b
独立地为h或c
1-c4烷基;例如c
1-c4烷基。
[0040]
在某一方案中,
[0041]r3a
和r
3b
独立地为h或c
1-c4烷基;例如c
1-c4烷基。
[0042]
在某一方案中,
[0043]ra1
、r
a3
和r
a5
独立地为h、卤素、cn、c
1-c6烷基-o-、被一个或多个r
1a
取代的c1‑ꢀ
c6烷基、或被一个或多个r
1b
取代的c
1-c6烷基-o-;
[0044]ra2
和r
a4
独立地为h、卤素;
[0045]
r4和r5独立地为h、c
1-c6烷基;或者,r4和r5与它们键合的碳一起形成3-7元环烷基;
[0046]
r6和r7独立地为h、-n(r
2ar2b
)或-n(r
3a
)-(c=o)-r
3b
;
[0047]
或者,r6和r7与它们键合的碳一起形成环b;环b为4-7元杂环烷基;
[0048]r1a
独立地为卤素;
[0049]r2a
、r
2b
、r
3a
和r
3b
独立地为h或c
1-c4烷基。
[0050]
在某一方案中,
[0051]ra1
、r
a3
和r
a5
独立地为h、卤素、cn、c
1-c6烷基-o-、被一个或多个r
1a
取代的c1‑ꢀ
c6烷基、或被一个或多个r
1b
取代的c
1-c6烷基-o-;
[0052]ra2
和r
a4
独立地为h、卤素;
[0053]
r4和r5独立地为h、c
1-c6烷基;或者,r4和r5与它们键合的碳一起形成3-7元环烷基;
[0054]
r6和r7与它们键合的碳一起形成环b;环b为4-7元杂环烷基;
[0055]r1a
独立地为卤素。
[0056]
在某一方案中,
[0057]ra1
、r
a2
、r
a3
、r
a4
和r
a5
独立地为h或卤素;
[0058]
为r4和r5独立地为c
1-c6烷基;
[0059]
或,r4和r5与它们键合的碳一起形成3-7元环烷基;
[0060]
r6和r7与它们键合的碳一起形成环b;环b为4-7元杂环烷基;
[0061]
例如环b为四氢吡喃基(例如);
[0062]
为
[0063]
在某一方案中,
[0064]ra1
、r
a2
、r
a3
、r
a4
和r
a5
独立地为h或卤素;
[0065]
为r4和r5独立地为c
1-c6烷基;或,r4和r5与它们键合的碳一起形成3-7元环烷基;
[0066]
r6和r7与它们键合的碳一起形成环b;环b为4-7元杂环烷基;
[0067]
例如环b为四氢吡喃基(例如);
[0068]
为
[0069]
在某一方案中,
[0070]
当r
a1
、r
a2
、r
a3
、r
a4
和r
a5
独立地为卤素时,所述的卤素为氟、氯或溴;例如氟或氯;又例如氟。
[0071]
在某一方案中,
[0072]
当r
a1
、r
a3
和r
a5
独立地为c
1-c6烷基、c
1-c6烷基-o-、或被一个或多个r
1a
取代的 c
1-c6烷基、或被一个或多个r
1b
取代的c
1-c6烷基-o-时,所述的c
1-c6烷基、c
1-c6烷基
ꢀ‑
o-、被一个或多个r
1a
取代的c
1-c6烷基和被一个或多个r
1b
取代的c
1-c6烷基-o-里的 c
1-c6烷基(例如甲
基、乙基、丙基、丁基、戊基或己基)独立地为c1~c4烷基(例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基);例如甲基或异丙基。
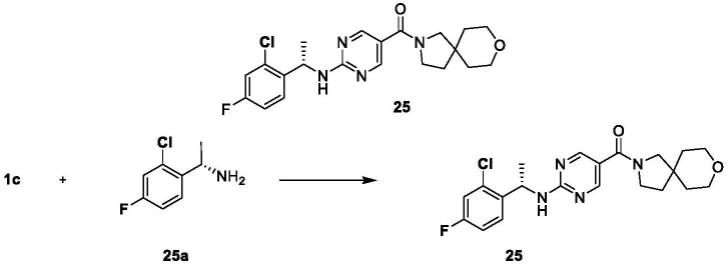
[0073]
在某一方案中,
[0074]
当r
a1
、r
a3
和r
a5
独立地为被一个或多个r
1a
取代的c
1-c6烷基或被一个或多个r
1b
取代的c
1-c6烷基-o-时,所述的取代基的个数为1、2、3、4或5个;例如1、2或3个;例如三氟甲基、三氟甲基-o-。
[0075]
在某一方案中,
[0076]
当r4和r5独立地为c
1-c6烷基、被一个或多个卤素取代的c
1-c6烷基时,所述的c1‑ꢀ
c6烷基和被一个或多个卤素取代的c
1-c6烷基里的c
1-c6烷基(例如甲基、乙基、丙基、丁基、戊基或己基)独立地为c1~c4烷基(例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基);例如甲基。
[0077]
在某一方案中,
[0078]
当r4和r5与它们键合的碳一起形成3-7元环烷基时,所述的3-7元环烷基独立地为环丁基、环戊基、环己基或环庚基;又例如环丙基。
[0079]
在某一方案中,
[0080]
当r6和r7独立地为卤素时,所述的卤素为氟、氯或溴;例如氟或氯。
[0081]
在某一方案中,
[0082]
当r6和r7独立地为c
1-c6烷基、或被一个或多个r
1c
取代的c
1-c6烷基时,所述的 c
1-c6烷基、和被一个或多个r
1c
取代的c
1-c6烷基里的c
1-c6烷基(例如甲基、乙基、丙基、丁基、戊基或己基)独立地为c1~c4烷基,例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基。
[0083]
在某一方案中,
[0084]
当环b为4-7元环烷基、或被一个或多个r
1d
取代的4-7元环烷基时,所述的4-7元环烷基、和被一个或多个r
1d
取代的4-7元环烷基里的4-7元环烷基独立地为环丁基、环戊基、环己基或环庚基。
[0085]
在某一方案中,
[0086]
当环b为4-7元杂环烷基、或被一个或多个r
1e
取代的4-7元杂环烷基时,所述的4
‑ꢀ
7元杂环烷基、和被一个或多个r
1e
取代的4-7元杂环烷基里的4-7元杂环烷基中,杂原子为n或o,杂原子个数为1个;例如四氢吡喃基,又例如
[0087]
在某一方案中,
[0088]
当r
1a
、r
1b
、r
1c
、r
1d
和r
1e
独立地为卤素、或被一个或多个卤素取代的c
1-c4烷基时,所述的卤素、和被一个或多个卤素取代的c
1-c4烷基里的卤素独立地为氟、氯或溴;例如氟或氯;又例如氟。
[0089]
在某一方案中,
[0090]
当r
1a
、r
1b
、r
1c
、r
1d
和r
1e
独立地为c
1-c4烷基、或被一个或多个卤素取代的c
1-c4烷基时,所述的c
1-c4烷基、和被一个或多个卤素取代的c
1-c4烷基里的c1~c4烷基独立地为甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基,例如甲基。
[0091]
在某一方案中,
[0092]
当r
2a
、r
2b
、r
3a
和r
3b
独立地为c
1-c4烷基时,所述的c1~c4烷基独立地为甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基,例如甲基。
[0093]
在某一方案中,
[0094]
为为为例如例如
[0095]
在某一方案中,
[0096]
为
[0097]
在某一方案中,
[0098]
为例如
[0099]
在某一方案中,
[0100]
所述的如式i所示的嘧啶甲酰胺类化合物选自如下任意结构,
[0101]
[0102][0103]
本发明中,所述的如式i所示的嘧啶甲酰胺类化合物或其药学上可接受的盐可具有一个或多个手性碳原子,因此可以分离得到光学纯度异构体,例如纯的对映异构体,或者外消旋体,或者混合异构体。可以通过本领域的分离方法来获得纯的单一异构体,如手性结晶成盐,或者手性制备柱分离得到。
[0104]
本发明中,所述的如式i所示的嘧啶甲酰胺类化合物或其药学上可接受的盐如存在立体异构体,则可以以单一的立体异构体或它们的混合物(例如外消旋体)的形式存在。术语“立体异构体”是指顺反异构体或旋光异构体。这些立体异构体可以通过不对称合成方法或手性分离法(包括但不限于薄层色谱、旋转色谱、柱色谱、气相色谱、高压液相色谱等)分离、纯化及富集,还可以通过与其它手性化合物成键(化学结合等)或成盐(物理结合等)等方式进行手性拆分获得。术语“单一的立体异构体”是指本发明化合物的一种立体异构体相对于该化合物的所有立体异构体的质量含量不低于95%。
[0105]
由此,在本说明书通篇中,本领域技术人员可对所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐中所述基团及其取代基进行选择,以提供稳定的如式i所示的嘧
啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或药学上可接受的盐,包括但不限于本发明的实施例中所述的化合物。
[0106]
本发明所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐可通过包括与化学领域公知方法相似的方法合成,其步骤和条件可参考本领域类似反应的步骤和条件,特别是根据本文说明进行合成。起始原料通常是来自商业来源,例如aldrich或可使用本领域技术人员公知的方法(通过scifinder、reaxys联机数据库得到)容易地制备。
[0107]
本发明中,所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐,也可以通过已制备得到的所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐,采用本领域常规方法,经外周修饰进而得到其他所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐。
[0108]
一般地,本发明的化合物可以通过本发明所描述的方法制备得到,除非有进一步的说明,其中取代基的定义如式i所示。下面的反应方案和实施例用于进一步举例说明本发明的内容。
[0109]
用于制备如式i中化合物的必要原料或试剂可以商购获得,或者通过本领域已知的合成方法制备。如下实验部分所描述的方法,可以制备游离碱或者其加酸所成盐的本发明的化合物。术语药学上可接受的盐指的是本文所定义的药学上可接受的盐,并且具有母体化合物所有的药学活性。药学上可接受的盐可以通过在有机碱的合适的有机溶剂中加入相应的酸,根据常规方法处理来制备药学上可接受的盐。
[0110]
成盐实例包括:与无机酸成盐,如盐酸、氢溴酸、硫酸、硝酸、磷酸;和有机酸所形成的盐,如醋酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、乙磺酸、富马酸、葡庚糖酸、谷氨酸、乙醇酸、羟基萘甲酸、2-羟基乙磺酸、乳酸、马来酸、苹果酸、丙二酸、扁桃酸、甲磺酸、黏糠酸、2-萘磺酸、丙酸、水杨酸、琥珀酸、酒石酸、对甲苯磺酸或三甲基乙酸。
[0111]
如式i所示的嘧啶甲酰胺类化合物可能具有一个或多个手性碳原子,因此可以分离得到光学纯度异构体,例如纯的对映异构体,或者外消旋体,或者混合异构体。可以通过本领域的分离方法来获得纯的单一异构体,如手性结晶成盐,或者手性制备柱分离得到。
[0112]
本专利所述的合成路线中所用的化学品,包括溶剂,试剂,催化剂以及保护基团,脱保护基团,保护基团包括叔丁氧基羰基(boc)。上述方法还可以另外包括在本文具体描述的步骤之前或之后的步骤,可以添加或除去合适的保护基团,以得到目标化合物。另外,各种合成步骤可以交替或顺次的进行以得到最终的目标产物。
[0113]
本发明提供了一种药物组合物,其包含如上所述的式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐,和,(一种或多种)药用辅料(例如可药用载体、稀释剂、媒介物或其它赋形剂)。所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐的用量可为治疗有效量。
[0114]
本发明还提供了一种所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐在制备vanin-1抑制剂中的应用。在所述的应用中,所述的vanin-1抑制剂可用于哺乳动物生物体内;也可用于生物体外,主要作为实验用途,例如:作为标准样或对照样提供比对,或按照本领域常规方法制成试剂盒,为vanin-1的抑制效果提供快速检测。如本文使用的术语“vanin-1(酶)的抑制剂”指结合vanin-1(酶)和降低产生的酶活性的化合物。
[0115]
本发明还提供了一种所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐在制备药物中的应用;所述的药物可为用于预防和/或治疗与vanin-1有关的疾病,或者,所述的药物可为用于预防和/或治疗自身免疫疾病、炎性疾病、变态反应性疾病、代谢疾病、基于感染的疾病、纤维变性疾病、心血管疾病、呼吸系统疾病、肾疾病、皮肤病学疾病、肝脏疾病、胃肠疾病、口腔疾病和造血疾病中的一种或多种;又例如克罗恩氏病、炎性肠病及溃疡性结肠炎的药物。所述的与vanin-1有关的疾病可包括自身免疫疾病、炎性疾病、变态反应性疾病、代谢疾病、基于感染的疾病、纤维变性疾病、心血管疾病、呼吸系统疾病、肾疾病、皮肤病学疾病、肝脏疾病、胃肠疾病、口腔疾病和造血疾病中的一种或多种;又例如炎性肠病、溃疡性结肠炎、克隆氏病、结肠直肠癌和胃炎。
[0116]
本发明的另一方面涉及一种预防和/或治疗vanin-1有关的疾病的方法,其包括向患者施用治疗有效剂量的所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐或包含其的药物组合物。例如治疗vanin-1酶的抑制介导的或另外地与vanin-1酶的抑制相关的疾病或病症。例如自身免疫疾病、炎性疾病、变态反应性疾病、代谢疾病、基于感染的疾病、纤维变性疾病、心血管疾病、呼吸系统疾病、肾疾病、皮肤病学疾病、肝脏疾病、胃肠疾病、口腔疾病和造血疾病中的一种或多种;又例如炎性肠病、溃疡性结肠炎、克隆氏病、结肠直肠癌和胃炎。
[0117]
本发明的另一方面涉及一种治疗预防和/或治疗自身免疫疾病、炎性疾病、变态反应性疾病、代谢疾病、基于感染的疾病、纤维变性疾病、心血管疾病、呼吸系统疾病、肾疾病、皮肤病学疾病、肝脏疾病、胃肠疾病、口腔疾病和造血疾病中的一种或多种(例如炎性肠病、溃疡性结肠炎、克隆氏病、结肠直肠癌和胃炎)的方法,其包括向患者施用治疗有效剂量的所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐或包含其的药物组合物。
[0118]
本发明的另一方面涉及一种用于抑制vanin-1的药物,其包括所述的如式i所示的嘧啶甲酰胺类化合物、或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其药学上可接受的盐或包含其的药物组合物。
[0119]
本发明的化合物可以用局部或全身给药,例如,用于肠内给药,比如直肠或口服用药,或用于肠胃外给药至哺乳动物(尤其指人)。本发明的化合物也可在肠胃外给药,例如,通过吸入式、注射或输液、如通过静脉内、动脉内、骨内、肌内、大脑内、脑室外、滑膜内、胸骨内、鞘内、病灶内、颅内、肿瘤内、皮内和皮下注射或输入。
[0120]
本发明所述化合物、药物组合物或药物的有效量取决于哺乳动物的种类、体重、年龄、个体状况、个体药代动力学参数、待治疗的疾病和给药方式。
[0121]
本发明所述化合物、药物组合物或药物的有效量可通过常规实验容易的测定,最有效和方便的给药途径以及最适当的制剂也可通过常规实验测定。
[0122]
所述的药用辅料可为药物生产领域中广泛采用的那些辅料。辅料主要用于提供一个安全、稳定和功能性的药物组合物,还可以提供方法,使受试者接受给药后活性成分以所期望速率溶出,或促进受试者接受组合物给药后活性成分得到有效吸收。所述的药用辅料可以是惰性填充剂,或者提供某种功能,例如稳定该组合物的整体ph值或防止组合物活性成分的降解。所述的药用辅料可以包括下列辅料中的一种或多种:粘合剂、助悬剂、乳化剂、稀释剂、填充剂、成粒剂、胶粘剂、崩解剂、润滑剂、抗粘着剂、助流剂、润湿剂、胶凝剂、吸收延迟剂、溶解抑制剂、增强剂、吸附剂、缓冲剂、螯合剂、防腐剂、着色剂、矫味剂和甜味剂。
[0123]
可作为药学上可接受辅料的物质包括,但并不限于,离子交换剂,铝,硬脂酸铝,卵磷脂,血清蛋白,如人血清蛋白,缓冲物质如磷酸盐,甘氨酸,山梨酸,山梨酸钾,饱和植物脂肪酸的部分甘油酯混合物,水,盐或电解质,如硫酸鱼精蛋白,磷酸氢二钠,磷酸氢钾,氯化钠,锌盐,胶体硅,三硅酸镁,聚乙烯吡咯烷酮,聚丙烯酸脂,蜡,聚乙烯
‑ꢀ
聚氧丙烯-阻断聚合体,羊毛脂,糖,如乳糖,葡萄糖和蔗糖;淀粉如玉米淀粉和土豆淀粉;纤维素和它的衍生物如羧甲基纤维素钠,乙基纤维素和乙酸纤维素;树胶粉;麦芽;明胶;滑石粉;辅料如可可豆脂和栓剂蜡状物;油如花生油,棉子油,红花油,麻油,橄榄油,玉米油和豆油;二醇类化合物,如丙二醇和聚乙二醇;酯类如乙基油酸酯和乙基月桂酸酯;琼脂;缓冲剂如氢氧化镁和氢氧化铝;海藻酸;无热原的水;等渗盐;林格(氏) 溶液;乙醇,磷酸缓冲溶液,和其他无毒的合适的润滑剂如月桂硫酸钠和硬脂酸镁,着色剂,释放剂,包衣衣料,甜味剂,调味剂和香料,防腐剂和抗氧化剂。
[0124]
本发明的药物组合物可根据公开的内容使用本领域技术人员已知的任何方法来制备。例如,常规混合、溶解、造粒、乳化、磨细、包封、包埋或冻干工艺。
[0125]
本发明的化合物的医药剂型可以以速释、控释、缓释或靶药物释放系统形式提供。例如,常用剂型包括溶液和悬浮液、(微)乳液、软膏、凝胶和贴片、脂质体、片剂、糖衣药丸、软壳或硬壳胶囊、栓剂、胚珠、植入物、非晶形或结晶粉末、气溶胶和冻干制剂。视所用的给药途径而定,可能需要特殊装置来施用或给予药物,例如注射器和针、吸入器、泵、注射笔、涂药器或专用瓶(specialflask)。药物剂型常常由药物、赋形剂和容器 /密封系统组成。可将一种或多种赋形剂(又称为非活性成分)添加到本发明的化合物中来改善或促进药物的制造、稳定性、给药和安全性,并且可提供获得所需药物释放曲线的方法。因此,添加到药物中的赋形剂类型可视各种因素而定,例如药物的物理和化学特性、给药途径和制备步骤。在该领域中存在药用赋形剂并且包括各种药典中所列的那些。(参见美国药典(u.s.pharmacopeia,usp)、日本药典(japanese pharmacopoeia,jp)、欧洲药典(european pharmacopoeia,ep)和英国药典(british pharmacopoeia,bp);美国食品与药品管理局(the u.s.food and drug administration,www.fda.gov)药物评价与研究中心 (centerfor drug evaluation and research,cedr)出版物,例如《非活性组分指南》(inactiveingredient guide,1996);ash和ash编写的《药物添加剂手册》(hand book of pharmaceuticaladditives,2002,联合信息资源公司(synapse information resources,inc.,endicott ny;etc.)。
[0126]
本发明化合物的药物剂型可通过本领域中熟知的任一种方法来制造,例如通过常
规混合、筛分、溶解、熔化、造粒、制造糖衣药丸、压片、悬浮、挤压、喷雾干燥、研磨、乳化、(纳米/微米级)囊封、包理或冻干工艺。
[0127]
本发明的药物组合物可以用局部或全身给药,例如,用于肠内给药,比如直肠或口服用药,或用于肠胃外给药至哺乳动物(尤其指人),并且包括根据本发明的化合物或其药学上可接受的盐作为活性成分的治疗有效量,连同药学上可接受的赋形剂,如药学上可接受的载体。活性成份的治疗有效量如上下文所定义,并且取决于哺乳动物的种类、体重、年龄、个体状况、个体药代动力学参数、待治疗的疾病和给药方式对于肠内给药,如口服药,本发明化合物可以配制成广泛的多种剂型。
[0128]
所述药物组合物和剂型可以包含一种或多种本发明的化合物或其一种或多种药学上可接受的盐作为活性组分。药学上可接受的载体可以是固体或液体。固体的形式的制剂包括粉剂、片剂、丸剂、锭剂、胶囊剂、扁嚢剂、栓剂和可分散的颗粒剂。固体载体可以还是作为稀释剂、调味剂、增溶剂、润滑剂、悬浮剂、粘合剂、防腐剂、片剂崩解剂或者包封材料的一种或多种物质。在粉剂中,载体通常是细碎的固体,其是与细碎的活性组分的混合物。在片剂中,活性组分通常与具有必要粘合能力的载体以合适的比例混合并按照所需的形状和尺寸压实。合适的载体包括但不限于碳酸镁、硬脂酸镁、滑石、糖、乳糖、果胶、糊精,淀粉、明胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡,可可脂等。活性化合物的制剂可以包括作为载体的包封材料,提供胶囊,其中有或没有载体的活性组分被与其结合的载体包围。
[0129]
适于口服给药的其它形式包括液体形式制剂,包括乳液、糖浆剂、酏剂、水溶液、水性悬浊液、或意图在使用前不久转化成液体形式制剂的固体形式制剂。乳液可以在溶液中制备,例如丙二醇水溶液中,或者可以含有乳化剂,如卵磷脂、脱水山梨糖醇单油酸酯或阿拉伯胶。水溶液可以通过将活性组分溶解在水中并加入合适的着色剂、香料、稳定剂和增稠剂来制备。水性混悬液可以通过将细小颗粒的活性成分用粘合剂如天然或合成胶、树脂、甲基纤维素、羧甲基纤维素和其它常用的悬浮剂分散在水中制备。固体形式的制剂包括溶液剂、混悬剂和乳液,除了活性组分外,还可以含有着色剂、香料、稳定剂、缓冲剂、人造和天然甜味剂、分散剂、增稠剂、增溶剂等。
[0130]
用于直肠给药的示例性组合包括栓剂,其可以包含例如适合的非刺激性赋形剂,例如可可脂、合成甘油酯或聚乙二醇,其在常温下是固体,但是在直肠腔中融化和/或溶解以释放药物。
[0131]
治疗有效剂量可首先使用本领域中熟知的各种方法来估算。用于动物研究的初始剂量可基于细胞培养测定中所确立的有效浓度。适合于人个体的剂量范围例如可使用从动物研究和细胞培养测定所获得的数据来确定。在某些实施方案中,可以将本发明的化合物制备为用于口服的药剂。
[0132]
药剂(例如本发明的化合物)的有效量或治疗有效量或剂量指的是引起个体症状改善或存活延长的药剂或化合物的量。所述分子的毒性和治疗功效可在细胞培养物或实验动物中通过标准医药程序来测定,例如通过测ld
50
(使群体50%致死的剂量)和ed
50
(对群体的50%治疗有效的剂量)。毒性作用与治疗作用的剂量比是治疗指数,可表示为ld
50
/ed
50
。优选显示高治疗指数的药剂。
[0133]
有效量或治疗有效量是将会引发研究人员、兽医、医生或其它临床医生所探求的组织、系统、动物或人类的生物或医学反应的化合物或医药组合物的量。剂量优选在包括极
小毒性或无毒性的ed
50
的循环浓度的范围内。剂量可在这个范围内变化,视所用的剂型和/或所用的给药途径而定。应根据本领域中已知的方法,考虑个体状况的特殊性来选择正确的制剂、给药途径、剂量和给药间隔时间。
[0134]
剂量和间隔时间可个别地加以调整以提供足以获得所需效果的活性部分的血浆水平;即最小有效浓度(minimal effective concentration,mec)。各化合物的mec将有所不同,但可以例如从体外(invitro)数据和动物实验估算。获得mec所必需的剂量将视个体特征和给药途径而定。在局部给药或选择性摄取的情况下,药物的有效局部浓度可能与血浆浓度无关。
[0135]
所施予的药剂或组合物的量可视各种因素而定,包括所治疗个体的性别、年龄和体重、病痛的严重性、给药方式和处方医师的判断。
[0136]
在需要时,本发明的组合物可以用含有一个或一个以上单位剂型(含有活性成分)的包装或分配装置提供。举例来说,所述包装或装置可包含金属或塑料箔(如发泡包装)或玻璃和橡皮塞,如在小瓶中。所述包装或分配装置可附有用药说明书。也可以制备包含在相容性医药载体中配制的本发明化合物的组合物,将其置于适当容器中,并且加上用于治疗指定病状的标签。
[0137]
除非另有规定,本文使用的所有技术术语和科学术语具有要求保护主题所属领域的标准含义。倘若对于某术语存在多个定义,则以本文定义为准。
[0138]
基团定义
[0139]
除非另外说明,应当应用本文所使用的下列定义。出于本发明的目的,化学元素与元素周期表cas版,和《化学和物理手册》,第75版,1994一致。此外,有机化学一般原理可参考"organic chemistry",thomas sorrell,university science books,sausalito:1999, 和"march's advanced organic chemistry”by michael b.smith and jerry march,john wiley& sons,new york:2007中的描述,其全部内容通过引用并入本文。
[0140]
在本说明书中,可由本领域技术人员选择基团及其取代基以提供稳定的结构部分和化合物。当通过从左向右书写的常规化学式描述取代基时,该取代基也同样包括从右向左书写结构式时所得到的在化学上等同的取代基。
[0141]
在本文中定义的某些化学基团前面通过简化符号来表示该基团中存在的碳原子总数。例如,c
1-c6烷基是指具有总共1、2、3、4、5或6个碳原子的如下文所定义的烷基。简化符号中的碳原子总数不包括可能存在于所述基团的取代基中的碳。
[0142]
在本文中,取代基中定义的数值范围如0至4、1-4、1至3等表明该范围内的整数,如1-6为1、2、3、4、5、6。
[0143]
除前述以外,当用于本技术的说明书及权利要求书中时,除非另外特别指明,否则以下术语具有如下所示的含义。
[0144]
术语“包括”为开放式表达,即包括本发明所指明的内容,但并不排除其他方面的内容。
[0145]
术语“被取代的”是指特定原子上的任意一个或多个氢原子被取代基取代,包括重氢和氢的变体,只要特定原子的价态是正常的并且取代后的化合物是稳定的。
[0146]
一般而言,术语“取代的”表示所给结构中的一个或多个氢原子被具体取代基所取代。进一步地,当该基团被1个以上所述取代基取代时,所述取代基之间是相互独立,即,所
述的1个以上的取代基可以是互不相同的,也可以是相同的。除非其他方面表明,一个取代基团可以在被取代基团的各个可取代的位置进行取代。当所给出的结构式中不只一个位置能被选自具体基团的一个或多个取代基所取代,那么取代基可以相同或不同地在各个位置取代。
[0147]
在本说明书的各部分,本发明公开化合物的取代基按照基团种类或范围公开。特别指出,本发明包括这些基团种类和范围的各个成员的每一个独立的次级组合。例如,术语“c1~c6烷基”或“c
1-6
烷基”特别指独立公开的甲基、乙基、c3烷基、c4烷基、c5烷基和 c6烷基;“c
1-4
烷基”特指独立公开的甲基、乙基、c3烷基(即丙基,包括正丙基和异丙基)、c4烷基(即丁基,包括正丁基、异丁基、仲丁基和叔丁基)。
[0148]
术语“卤素”选自于f,cl,br或i,尤其指f或cl。
[0149]
在本技术中,作为基团或是其它基团的一部分,术语“烷基”是指饱和脂肪族烃基团,其为包含1至12个碳原子的直链或支链基团,优选含有1至6个碳原子的烷基直链或支链的。通式c
nh2n 1
。术语“c
1-c6烷基”是指烷基部分包含1,2,3,4,5或6个碳原子。在某一实施方案中,所述的“烷基”是指c
1-c6烷基。在某一实施方案中,所述的“烷基”是指c
1-c4烷基。
[0150]
含有1至6个碳原子的低级烷基,非限制性实施例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、 2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、 1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基等。含有1至12个碳原子的非限制性实例包括上述含有1至6个碳原子的低级烷基的实例,以及2,4-二甲基戊基、2,2-二甲基戊基、3,3-二甲基戊基、2-乙基戊基、3-乙基戊基、正辛基、2,3-二甲基己基、2,4-二甲基己基、2,5-二甲基己基、2,2-二甲基己基、3, 3-二甲基己基、4,4-二甲基己基、2-乙基己基、3-乙基己基、4-乙基己基、2-甲基-2-乙基戊基、2-甲基-3-乙基戊基、正壬基、2-甲基-2-乙基己基、2-甲基-3-乙基己基、2,2-二乙基戊基、正奎基、3,3-二乙基己基、2,2-二乙基己基,及其各种支链异构体等。
[0151]
在本技术中,作为基团或是其它基团的一部分,除非另有规定,术语“环烷基”意指仅由碳原子和氢原子组成的饱和的单环、多环或者桥接碳环取代基,且其可经由任何适宜的碳原子通过单键与分子的其余部分连接;当为多环时,可为并环连接或螺环连接(即,碳原子上的两个偕氢被亚烷基取代)的桥环体系或螺环体系。环烷基取代基可以经任何适宜的碳原子连接在中心分子上。在一些实施例中,具有3-10个碳原子的环可以表示为 c
3-c
10
环烷基。在一些实施例中,c3~c6的环烷基包括环丙基(c3)、环丁基(c4)、环戊基(c5)及环己基(c6)。在一些实施例中,c3~c
10
的环烷基的实例包括上述c3~c6环烷基基团连同环庚基(c7)、环辛基(c8)、环壬基(c9)及环癸基(c
10
)。
[0152]
在本技术中,作为基团或是其它基团的一部分,术语“杂环烷基”意指由2-6个碳原子以及1-4个选自氮、氧和硫的杂原子组成的稳定的3元至7元饱和环状基团。示例性3
‑ꢀ
元杂环基基团包括但不限于,氮杂环丙基、环氧乙烷基以及硫杂环丙烷基,或者其立体异构体;示例性4-元杂环基基团包括但不限于,氮杂环丁烷基,环氧丙烷基,硫杂环丁烷基,或者其同分异构体和立体异构体;示例性5-元杂环基基团包括但不限于,四氢呋喃基,四氢噻吩基,吡咯烷基,噻唑烷基,异噻唑烷基,噁唑烷基,异噁唑烷基,咪唑烷基,吡唑烷基,二氧戊
环基,氧杂硫呋喃基,二硫呋喃基,或者其同分异构体和立体异构体。示例性6-元杂环基基团包括但不限于,哌啶基,四氢吡喃基,硫化环戊烷基,吗啉基,硫代吗啉基,二噻烷基,二噁烷基,哌嗪基,三嗪烷基,或者其同分异构体和立体异构体;示例性7-元杂环基基团包括但不限于,氮杂环庚烷基,氧杂环庚烷基,硫杂环庚烷基,以及二氮杂环庚基,或者其同分异构体和立体异构体。在某一方案中,“杂环烷基”为c2~c5杂环烷基,其中杂原子选自n、o和s中的一种或多种,杂原子数为1、2或3 个。
[0153]
本文所用术语“部分”、“结构部分”、“化学部分”、“基团”、“化学基团”是指分子中的特定片段或官能团。化学部分通常被认为是嵌入或附加到分子上的化学实体。
[0154]
当所列举的取代基中没有指明其通过哪一个原子连接到化学结构通式中包括但未具体提及的化合物时,这种取代基可以通过其任何原子相键合。取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。
[0155]
在本发明的各部分,描述了连接取代基。当该结构清楚地需要连接基团时,针对该基团所列举的马库什变量应理解为连接基团。例如,如果该结构需要连接基团并且针对该变量的马库什基团定义列举了“烷基”,则应该理解,该“烷基”代表连接的亚烷基基团。
[0156]
在一些具体的结构中,当烷基基团清楚地表示为连接基团时,则该烷基基团代表连接的亚烷基基团,例如,基团“卤代-c1~c6烷基”中的c
1-c6烷基应当理解为c1~c6亚烷基。
[0157]
术语“亚烷基”表示从饱和的直链或支链烃基中去掉两个氢原子所得到的饱和的二价烃基基团。亚烷基基团的实例包括亚甲基(-ch
2-),亚乙基{包括-ch2ch
2-或-ch(ch3)-},亚异丙基{包括-ch(ch3)ch
2-或-c(ch3)
2-}等等。
[0158]
除非另有规定,本文使用的所有技术术语和科学术语具有要求保护主题所属领域的标准含义。倘若对于某术语存在多个定义,则以本文定义为准。
[0159]
应该理解,在本发明中使用的单数形式,如“一种”,包括复数指代,除非另有规定。此外,术语“包括”是开放性限定并非封闭式,即包括本发明所指明的内容,但并不排除其他方面的内容。
[0160]
除非另有说明,本发明采用质谱、元素分析的传统方法,各步骤和条件可参照本领域常规的操作步骤和条件。
[0161]
除非另有指明,本发明采用分析化学、有机合成化学和光学的标准命名及标准实验室步骤和技术。在某些情况下,标准技术被用于化学合成、化学分析、发光器件性能检测。
[0162]
另外,需要说明的是,除非以其他方式明确指出,在本发明中所采用的描述方式
“…
独立地为”应做广义理解,是指所描述的各个个体之间是相互独立的,可以独立地为相同或不同的具体基团。更详细地,描述方式
“…
独立地为”既可以是指在不同基团中,相同符号之间所表达的具体选项之间互相不影响;也可以表示在相同的基团中,相同符号之间所表达的具体选项之间互相不影响。
[0163]
本领域技术人员可以理解,根据本领域中使用的惯例,本技术描述基团的结构式中所使用的是指,相应的基团通过该位点与化合物中的其它片段、基团进行连接。
[0164]“药学上可接受的”是指可用于制备药物组合的情形,通常是安全的和无毒的,且非生物学以及其它方面所不期望,并且包括对于兽用和用于人的药物用途的可接受的情形。
[0165]
术语“赋形剂”是指药学上可接受的化学物质,例如药学领域的普通技术人员已知
的用于帮助给予药用的试剂。它是可以用于制备药物组分的化合物,通常是安全的、无毒的,且是生物学或者其它方面所不可期望的,其包括对于兽用和人用药物可接受的赋形剂。通常的赋形剂包括粘合剂、表面活性剂、稀释剂、崩解剂和润滑剂。
[0166]
术语“有效治理量”是指当给予受试者来治疗疾病状态时足以实现疾病状态的这种治理的所用化合物的量。“有效治理量”将根据化合物、所治疗的疾病状态、所治疗疾病的严重程度、受试者的年龄和相对健康、给药途径和方式、主治医疗或兽医的判断等而变化。
[0167]
术语哺乳动物是指人或任何哺乳动物,如灵长类动物、农场动物、宠物动物或者实验动物。这些动物的实例有猴、母牛、羊、马、猪、狗、猫、兔、小鼠和大鼠等。哺乳动物优选于人。
[0168]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0169]
本发明所用试剂和原料均市售可得。
[0170]
本发明的积极进步效果在于:提供了一种嘧啶甲酰胺类化合物,可以用作vanin酶、特别是vanin-1抑制剂;其可用于制备预防和/或治疗克罗恩氏病、溃疡性结肠炎及炎性肠病。
具体实施方式
[0171]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0172]
实施例1
[0173][0174][0175]
第一步
[0176]
在-10℃和氮气保护的条件下,向化合物1a(25.40g),化合物1b(31.00g)和三乙胺(61.00g)的乙腈(250ml)溶液中缓慢滴加t3p(丙基磷酸酐)(254.00g)。滴加完毕后,维持体系在-5℃反应3h。反应结束。向反应液中加入300ml水淬灭反应,浓缩除去有机溶剂,残留物在5℃搅拌1h,有固体析出,过滤,滤饼用水(100ml x 1)洗涤,干燥,得化合物1c(42.00g),收率93%。
[0177]1h nmr(400mhz,cdcl3)δ8.80(s,2h),3.81-3.57(m,7h),3.36(s,1h),1.93(td,j= 14.6,7.2hz,2h),1.66(t,j=5.4hz,2h),1.58(dd,j=11.1,4.6hz,2h).
[0178]
第二步
[0179]
将化合物1d(400mg,2.82mmol),1c(792.5mg,2.82mmol)和n,n-二异丙基乙胺(2ml)的1,4-二氧六环(30ml)溶液加热到100℃反应过夜。反应完毕,冷却到室温,减压浓缩,得到残留物进行硅胶柱(甲醇/二氯甲烷=0-100%)纯化,得到灰白色固体化合物1(356mg,收率:33%)。
[0180]1h nmr(400mhz,dmso-d6)δ8.55(s,2h),8.23(brs,1h),7.29-7.47(m,4h),4.67-4.61 (m,2h),3.53-3.64(m,8h),1.80(m,2h),1.45-1.55(m,4h).
[0181]
lcms(esi),[m h]
=387.1
[0182]
实施例2
[0183][0184]
第一步
[0185]
按照实施例1的方法,由化合物1c和2a得到化合物2(10mg,收率:4%)。
[0186]1h nmr(400mhz,dmso-d6)δ10.7(brs,1h),9.04-9.06(m,2h),8.30(m,1h),7.79(m 2h),7.65(m,1h),5.33(s,2h),3.44-3.65(m,8h),1.83-1.88(m,2h),1.46-1.49(m,4h).
[0187]
lcms(esi),[m h]
=378.1
[0188]
实施例3
[0189][0190]
第一步
[0191]
按照实施例1的方法,由化合物1c和3a得到化合物3(150mg,收率:36%)。
[0192]1h nmr(400mhz,dmso-d6)δ8.50(s,2h),8.27-8.29(m,1h),7.49-7.43(m,1h),7.25
‑ꢀ
7.27(m,1h),7.11-7.16(m,2h),5.42(m,1h),3.51-3.59(m,6h),3.42(m,2h),1.76-1.78(m, 2h),1.44-1.52(m,7h).
[0193]
lcms(esi),[m h]
=385.2
[0194]
实施例4
[0195][0196]
第一步
[0197]
按照实施例1的方法,由化合物1c和4a得到化合物4(105mg,收率:25%)。
[0198]1h nmr(400mhz,dmso-d6)δ8.49(s,2h),8.27-8.29(m,1h),7.49-7.43(m,1h),7.25
‑ꢀ
7.27(m,1h),7.11-7.16(m,2h),5.42(m,1h),3.40-3.59(m,8h),1.76-1.78(m,2h),1.23-1.52 (m,7h).
[0199]
lcms(esi),[m h]
=385.2
[0200]
实施例5
[0201][0202]
第一步
[0203]
按照实施例1的方法,由化合物1c和5a得到化合物5(57mg,收率:45%)。
[0204]1h nmr(400mhz,dmso-d6)δ8.52(s,2h),8.21(s,1h),7.31(d,j=4.4hz,4h), 7.26-7.17(m,1h),4.55(m,2h),3.68-3.35(m,8h),1.78(m,2h),1.53-1.45(m,4h).
[0205]
lcms(esi),[m h]
=353.2
[0206]
实施例6
[0207][0208]
第一步
[0209]
按照实施例1的方法,由化合物1c和6a得到化合物6(27mg,收率:30%)。
[0210]1h nmr(400mhz,dmso-d6)δ8.48(s,2h),8.22(m,1h),7.38(m,2h),7.30(m,2h), 7.19(m,1h),5.16(m,1h),3.66-3.34(m,8h),1.77(m,2h),1.52(s,1h),1.45(m,5h),1.24
(s, 1h).
[0211]
lcms(esi),[m h]
=367.2
[0212]
实施例7
[0213][0214]
第一步
[0215]
按照实施例1的方法,由化合物1c和7a得到化合物7(37mg,收率:28%)。
[0216]1h nmr(400mhz,dmso-d6)δ8.52(m,2h),8.22(s,1h),7.45-7.27(m,2h),7.21
‑ꢀ
6.95(m,2h),4.52(m,2h),3.70-3.34(m,8h),1.78(m,2h),1.49(m,4h).
[0217]
lcms(esi),[m h]
=371.1
[0218]
实施例8
[0219][0220]
第一步
[0221]
按照实施例1的方法,由化合物1c和8a得到化合物8(29mg,收率:30%)。
[0222]1h nmr(400mhz,dmso-d6)δ8.53(m,2h),8.19(s,1h),7.44-7.23(m,2h),7.23
‑ꢀ
7.07(m,2h),4.59(m,2h),3.69-3.35(m,8h),1.78(m,2h),1.54-1.44(m,4h).
[0223]
lcms(esi),[m h]
=371.1
[0224]
实施例9
[0225][0226]
第一步
[0227]
按照实施例1的方法,由化合物1c和9a得到化合物9(43mg,收率:47%)。
[0228]1h nmr(400mhz,dmso-d6)δ8.56(s,2h),7.95(s,1h),7.60(m,3h),4.65(m,2h), 3.72-3.41(m,7h),1.80(s,2h),1.50(m,4h),1.24(s,1h).
[0229]
lcms(esi),[m h]
=439.1
[0230]
实施例10
[0231][0232]
第一步
[0233]
按照实施例1的方法,由化合物1c和10a得到化合物10(53mg,收率:64%)。
[0234]1h nmr(400mhz,dmso-d6)δ8.53(s,2h),8.03(s,1h),7.38(m,1h),7.08(m,2h), 4.59(m,2h),3.81-3.38(m,7h),1.79(s,2h),1.49(m,4h).
[0235]
lcms(esi),[m h]
=389.2
[0236]
实施例11
[0237][0238]
第一步
[0239]
按照实施例1的方法,由化合物1c和11a得到化合物11(51mg,收率:56%)。
[0240]1h nmr(400mhz,dmso-d6)δ8.52(m,2h),8.22(s,1h),7.45-7.27(m,2h),7.21
‑ꢀ
6.95(m,2h),4.52(m,2h),3.70-3.34(m,7h),1.78(m,2h),1.49(m,4h).
[0241]
lcms(esi),[m h]
=421.2.
[0242]
实施例12
[0243][0244]
第一步
[0245]
按照实施例1的方法,由化合物1c和12a得到化合物12(55mg,收率:66%)。
[0246]1h nmr(400mhz,dmso-d6)δ8.53(s,2h),8.19(s,1h),7.39(m,1h),7.21(m,1h), 7.04(m,1h),4.55(m,2h),3.67
–
3.35(m,8h),1.78(m,2h),1.49(m,4h).
[0247]
lcms(esi),[m h]
=389.1
[0248]
实施例13
[0249][0250]
第一步
[0251]
按照实施例1的方法,由化合物1c和13a得到化合物13(58mg,收率:62%)。
[0252]1h nmr(400mhz,dmso-d6)δ8.54(s,2h),8.20(s,1h),7.53-7.28(m,4h),4.64(m, 2h),3.73-3.39(m,8h),1.80(s,2h),1.50(m,4h).
[0253]
lcms(esi),[m h]
=437.1
[0254]
实施例14
[0255][0256][0257]
第一步
[0258]
按照实施例1的方法,由化合物1c和14a得到化合物14(29mg,收率:31%)。
[0259]1h nmr(400mhz,dmso-d6)δ8.52(s,2h),8.25(s,1h),7.43(m,2h),7.30(m,2h), 4.57(m,2h),3.59-3.45(m,8h),1.78(s,2h),1.55-1.43(m,4h).
[0260]
lcms(esi),[m h]
=437.1
[0261]
实施例15
[0262][0263]
第一步
[0264]
按照实施例1的方法,由化合物1c和15a得到化合物15(83mg,收率:98%)。
[0265]1h nmr(400mhz,dmso-d6)δ8.49(m,3h),7.62(m,1h),7.30-7.19(m,1h),7.10(m, 2h),3.52(m,8h),1.78(s,2h),1.49(m,4h),1.26(m,2h),1.18(m,2h).
[0266]
lcms(esi),[m h]
=397.2
[0267]
实施例16
[0268][0269]
第一步
[0270]
按照实施例1的方法,由化合物1c和16a得到化合物16(57mg,收率:71%)。
[0271]1h nmr(400mhz,dmso-d6)δ8.53(s,2h),8.00(s,1h),7.29-7.20(m,1h),7.18(s, 1h),6.99(m,1h),6.89(m,1h),4.53(m,2h),3.84(m,3h),3.70-3.39(m,8h),1.80(s,2h), 1.51(m,4h).
[0272]
lcms(esi),[m h]
=383.2
[0273]
实施例17
[0274][0275]
第一步
[0276]
按照实施例1的方法,由化合物1c和17a得到化合物17(61mg,收率:70%)。
[0277]1h nmr(400mhz,dmso-d6)δ8.51(s,2h),8.12(s,1h),7.21(m,2h),6.87-6.79(m, 2h),4.55(m,1h),4.46(m,2h),3.66-3.43(m,8h),1.79(s,2h),1.49(m,4h),1.23(m,6h).
[0278]
lcms(esi),[m h]
=411.2
[0279]
实施例18
[0280][0281]
按照实施例1的方法,由化合物1c和18a得到化合物18(15mg,收率:17%)。
[0282]1h nmr(400mhz,dmso-d6)δ8.55(m,3h),7.34-7.09(m,4h),3.68-3.48(m,8h), 1.78(m,2h),1.49(m,4h),1.30-1.24(m,3h),1.19(m,1h).
[0283]
lcms(esi),[m h]
=413.1
[0284]
实施例19
[0285][0286]
按照实施例1的方法,由化合物1c和19a得到化合物19(46mg)。
[0287]1h nmr(400mhz,dmso-d6)δ8.51(s,2h),8.33-8.26(m,1h),7.53-7.46(m,1h), 7.20-7.16(m,1h),7.10-7.02(m,1h),5.38(brs,1h),3.66-3.40(m,8h),1.83-1.70(m,2h), 1.58-1.35(m,7h).
[0288]
lcms(esi),[m h]
=403.2
[0289]
实施例20
[0290][0291]
按照实施例1的方法,由化合物1c和20a得到化合物20(55mg)。
[0292]1h nmr(400mhz,dmso-d6)δ8.51(s,2h),8.33-8.26(m,1h),7.53-7.46(m,1h), 7.20-7.16(m,1h),7.10-7.02(m,1h),5.38(brs,1h),3.66-3.40(m,8h),1.83-1.70(m,2h), 1.58-1.35(m,7h).
[0293]
lcms(esi),[m h]
=403.2
[0294]
实施例21
[0295][0296]
按照实施例1的方法,由化合物1c和21a得到化合物21(50mg)。
[0297]1h nmr(400mhz,dmso-d6)δ8.51(s,2h),8.34-8.30(m,1h),7.49-7.43(m,1h), 7.40-7.37(m,1h),7.29-7.25(m,1h),5.36(brs,1h),3.66-3.40(m,8h),1.83-1.70(m,2h), 1.58-1.35(m,7h).
[0298]
lcms(esi),[m h]
=419.1
[0299]
实施例22
[0300][0301]
按照实施例1的方法,由化合物1c和22a得到化合物22(16mg)。
[0302]1h nmr(400mhz,dmso-d6)δ8.48(s,2h),8.17-8.16(m,1h),7.12-7.08(m,2h), 5.36-5.33(m,1h),3.60-3.37(m,8h),1.78-1.76(m,2h),1.55-1.37(m,7h).
[0303]
lcms(esi),[m h]
=421.2
[0304]
实施例23
[0305][0306]
按照实施例1的方法,由化合物1c和23a得到化合物23(48mg)。
[0307]1h nmr(400mhz,dmso-d6)δ8.48(s,2h),8.17-8.16(m,1h),7.12-7.08(m,2h), 5.36-5.33(m,1h),3.60-3.36(m,8h),1.78-1.75(m,2h),1.56-1.37(m,7h).
[0308]
lcms(esi),[m h]
=421.2
[0309]
实施例24
[0310][0311]
按照实施例1的方法,由化合物1c和24a得到化合物24(32mg)。
[0312]1h nmr(400mhz,dmso-d6)δ8.50(s,2h),8.40-8.35(m,1h),7.57-7.53(m,1h), 7.40-7.37(m,1h),7.23-7.18(m,1h),5.43-5.39(m,1h),3.61-3.40(m,8h),1.81-1.74(m,2h), 1.56-1.37(m,7h).
[0313]
lcms(esi),[m h]
=419.1
[0314]
实施例25
[0315][0316]
按照实施例1的方法,由化合物1c和25a得到化合物25(31mg)。
[0317]1h nmr(400mhz,dmso-d6)δ8.49(s,2h),8.38-8.34(m,1h),7.55(brs,1h),7.39
‑ꢀ
7.37(m,1h),7.22-7.17(m,1h),5.41(brs,1h),3.60-3.36(m,8h),1.80-1.74(m,2h),1.56
‑ꢀ
1.37(m,7h).
[0318]
lcms(esi),[m h]
=419.1
[0319]
实施例26
[0320][0321]
按照实施例1的方法,由化合物1c和26a得到化合物26(43mg)。
[0322]1h nmr(400mhz,dmso-d6)δ8.50-8.35(m,3h),7.91-7.87(m,1h),7.58-7.51(m, 2h),5.50-5.39(m,1h),3.61-3.40(m,8h),1.81-1.72(m,2h),1.56-1.37(m,7h).
[0323]
lcms(esi),[m h]
=453.2.
[0324]
实施例27
[0325][0326]
按照实施例1的方法,由化合物1c和27a得到化合物27(67mg)。
[0327]1h nmr(400mhz,dmso-d6)δ8.50-8.35(m,3h),7.57-7.55(m,1h),7.30-7.15(m, 3h),5.50-5.39(m,1h),3.63-3.40(m,8h),1.81-1.72(m,2h),1.56-1.37(m,7h).
[0328]
lcms(esi),[m h]
=467.1.
[0329]
实施例28
[0330][0331]
按照实施例1的方法,由化合物1c和28a得到化合物28(41mg)。
[0332]1h nmr(400mhz,dmso-d6)δ8.55-8.35(m,3h),7.83(s,1h),7.72(brs,2h),5.50
‑ꢀ
5.39(m,1h),3.61-3.40(m,8h),1.81-1.71(m,2h),1.56-1.37(m,7h).
[0333]
lcms(esi),[m h]
=469.1.
[0334]
实施例29
[0335][0336]
按照实施例1的方法,由化合物1c和29a得到化合物29(33mg)。
[0337]1h nmr(400mhz,dmso-d6)δ8.55-8.35(m,3h),7.82(s,1h),7.72(brs,2h),5.50
‑ꢀ
5.39(m,1h),3.61-3.40(m,8h),1.81-1.71(m,2h),1.56-1.37(m,7h).
[0338]
lcms(esi),[m h]
=469.1.
[0339]
实施例30
[0340][0341]
按照实施例1的方法,由化合物1c和30a得到化合物30(16mg)。
[0342]1h nmr(400mhz,dmso-d6)δ8.43-8.32(m,3h),7.75-7.55(m,3h),5.32(brs,1h), 5.55-5.30(m,8h),1.69(brs,2h),1.45-1.35(m,7h).
[0343]
lcms(esi),[m h]
=410.2.
[0344]
实施例31
[0345][0346]
按照实施例1的方法,由化合物1c和31a得到化合物31(52mg)。
[0347]1h nmr(400mhz,dmso-d6)δ8.55-8.35(m,3h),7.83(s,1h),7.72(brs,2h),5.50
‑ꢀ
5.39(m,1h),3.61-3.40(m,8h),1.81-1.71(m,2h),1.56-1.37(m,7h).
[0348]
lcms(esi),[m h]
=425.1.
[0349]
实施例32
[0350][0351]
按照实施例1的方法,由化合物1c和32a得到化合物32(52mg)。
[0352]1h nmr(400mhz,dmso-d6)δ8.50(brs,2h),8.35-8.33(m,1h),7.28-7.16(m,3h), 5.43-5.40(m,1h),3.61-3.40(m,8h),1.78-1.76(m,2h),1.56-1.37(m,7h).
[0353]
lcms(esi),[m h]
=403.2.
[0354]
实施例33
[0355][0356]
按照实施例1的方法,由化合物1c和33a得到化合物33(28mg)。
[0357]1h nmr(400mhz,dmso-d6)δ8.50(brs,2h),8.35-8.33(m,1h),7.28-7.16(m,3h), 5.43-5.40(m,1h),3.65-3.48(m,8h),1.78-1.76(m,2h),1.56-1.37(m,7h).
[0358]
lcms(esi),[m h]
=403.2.
[0359]
实施例34
[0360][0361][0362]
第一步
[0363]
将化合物1a(2g.12.7mmol溶于dcm(25ml)中,冰浴下向其中滴入1.6ml草酰氯(2.42g,19mmol)和6滴dmf,撤去冰浴,自然升至室温并继续搅拌2h。反应结束后,减压除去溶剂得到粗产品34a(2.1g)。
[0364]
第二步
[0365]
将化合物34a(2.1g,12mmol),化合物34b(1.9g,11mmol)溶于乙腈(25ml) 中,在冰浴下向其中滴入t3p(20ml,60%in ea,10g,31.25mmol)。滴加完毕后,自然升至室温并继续搅拌2h。反应结束后,将反应混合物用水(50ml)淬灭,用二氯甲烷(20ml
×
3)萃取,无水硫酸钠干燥,真空浓缩,经硅胶柱(meoh/dcm=0-100)纯化得到化合物34c(600mg)。
[0366]
第三步
[0367]
按照实施例1的方法,由化合物34c和34d得到化合物34(134mg)。
[0368]1h nmr(400mhz,dmso-d6)δ8.51(brs,2h),8.36-8.33(m,1h),7.53-7.46(m,1h), 7.22-7.17(m,1h),7.08-7.04(m,1h),5.38(brs,1h),5.03-4.48(m,1h),3.68-3.35(m,4h), 2.86-2.71(m,3h),2.15-1.91(m,5h),1.46-1.44(m,3h)。
[0369]
lcms(esi),[m h]
=404.1.
[0370]
实施例35
[0371][0372]
按照实施例1的方法,由化合物34c和35a得到化合物35(134mg)。
[0373]1h nmr(400mhz,dmso-d6)δ8.51(brs,2h),8.36-8.33(m,1h),7.53-7.46(m,1h), 7.22-7.17(m,1h),7.08-7.04(m,1h),5.38(brs,1h),5.03-4.46(m,1h),3.68-3.35(m,4h), 2.86-2.71(m,3h),2.15-1.91(m,5h),1.46-1.44(m,3h).
[0374]
lcms(esi),[m h]
=404.1.
[0375]
实施例36
[0376][0377]
按照实施例1的方法,由化合物34c和36a得到化合物36(12mg)。
[0378]1h nmr(400mhz,dmso-d6)δ8.50(brs,2h),8.36-8.33(m,1h),7.47-7.25(m,3h), 5.36-5.32(m,1h),4.94-4.47(m,1h),3.69-3.40(m,4h),2.86-2.71(m,3h),2.03-1.83(m,5h), 1.46-1.41(m,3h).
[0379]
lcms(esi),[m h]
=420.1.
[0380]
实施例37
[0381][0382]
按照实施例1的方法,由化合物34c和37a得到化合物37(12mg)。
[0383]1h nmr(400mhz,dmso-d6)δ8.50(brs,2h),8.36-8.32(m,1h),7.47-7.24(m,3h), 5.36-5.32(m,1h),4.94-4.47(m,1h),3.69-3.40(m,4h),2.86-2.71(m,3h),2.03-1.83(m,
5h), 1.46-1.41(m,3h).
[0384]
lcms(esi),[m h]
=420.1.
[0385]
实施例38
[0386][0387]
按照实施例1的方法,由化合物34c和38a得到化合物38(41mg)。
[0388]1h nmr(400mhz,dmso-d6)δ8.47(brs,2h),8.02-7.96(m,1h),7.57-7.50(m,3h), 5.50-5.46(m,1h),3.67-3.51(m,4h),2.86-2.71(m,4h),2.03-1.83(m,5h),1.58-1.56(m,3h).
[0389]
lcms(esi),[m h]
=454.1.
[0390]
实施例39
[0391][0392]
按照实施例1的方法,由化合物34c和39a得到化合物39(100mg)。
[0393]1h nmr(400mhz,dmso-d6)δ8.49(s,2h),8.40(s,1h),7.57-7.52(m,1h),7.41
‑ꢀ
7.55(m,1h),7.23-7.18(m,1h),5.45-5.36(m,1h),3.66-3.59(m,4h),2.85(s,1h),2.71(s, 1h),2.05-1.97(m,6h),1.45-1.40(m,3h),1.23(s,1h).
[0394]
lcms(esi),[m h]
=420.1.
[0395]
实施例40
[0396][0397]
按照实施例1的方法,由化合物34c和40a得到化合物40(100mg)。
[0398]1h nmr(400mhz,dmso-d6)δ8.51(s,2h),8.39-8.35(m,1h),7.28-7.25(m,2h), 7.20-7.12(m,1h),5.43-5.40(m,1h),3.64-3.46(m,4h),2.85(s,1h),2.71(s,1h),2.05-1.94 (m,6h),1.50-1.45(m,3h),1.24(s,1h).
[0399]
lcms(esi),[m h]
=404.2.
[0400]
实施例41
[0401][0402]
按照实施例1的方法,由化合物34c和41a得到化合物41(1800mg)。
[0403]1h nmr(400mhz,dmso-d6)δ8.59-8.35(m,3h),7.69-7.58(m,3h),5.42-5.38(m, 1h),5.02-4.39(m,1h),4.46(brs,1h),3.69-3.44(m,4h),3.00-2.85(m,3h),2.03-1.85(m,4h), 1.48-1.42(m,3h).
[0404]
lcms(esi),[m h]
=454.2.
[0405]
化合物41经手性拆分得到两个对映异构体41-p1和41
‑‑
p2。
[0406]
色谱条件如下:
[0407]
色谱柱:chiralpak ig(5um,30*250mm i.d)
[0408]
流速:80ml/min
[0409]
波长:220nm
[0410]
柱温:38℃
[0411]
流动相:a:co2,b:methanol
[0412]
41-p2手性合成
[0413][0414]
第一步
[0415]
将化合物a(50g,0.26mol),化合物b(38g,0.31mol)和钛酸四乙酯(120g,0.52 mol)的thf(500ml)溶液加热至40℃并搅拌2h。反应结束后,冷却,向体系中加入 300ml水,剧烈搅拌0.5小时,过滤,滤饼依次用水(100mlx1)和乙酸乙酯(100mlx1) 洗涤,滤液用乙酸乙酯萃取(300mlx3),合并有机相并干燥,浓缩得粗产物c(72g)。
[0416]
第二步
[0417]
将化合物c(72g,0.25mol)溶于2-甲基四氢呋喃(750ml)中,在-78℃下缓慢滴加甲基溴化镁(3m in 2-me-thf,420ml)。滴加完后,反应体系于-78℃继续搅拌2h。反应结束,加入饱和氯化铵水溶液(300ml)淬灭反应。将淬灭后的混合物减压除去绝大部分2-甲基四氢呋喃,用乙酸乙酯萃取(500mlx3),合并有机相并干燥,浓缩,得到粗产物d(78g)。
[0418]
第三步
[0419]
在0℃下,向化合物d(40g,0.129mol)的乙酸乙酯(50ml)溶液中缓慢加入盐酸乙酸乙酯溶液(3m,400ml)。滴加完毕后,反应液继续室温搅拌2h。反应结束,过滤,得到白色固体。将此白色固体分散到naoh溶液(4n,100ml)中,乙酸乙酯(300mlx2) 萃取,干燥,浓缩得粗产物e(16g,chiral hplc:78%ee)。该粗品经手性拆分后得目标产物e(9.6g,chiral hplc:100%ee).
[0420]
第四步
[0421]
按照实施例41的合成方法,由化合物e(8.7g,0.038mol)和化合物34c(9.7g, 0.034mol)得到目标产物41-p2(9.7g)。
[0422]1h nmr(400mhz,cd3od)δ8.50(s,2h),7.62-7.57(m,1h),7.45-7.34(m,2h),5.54
‑ꢀ
5.40(m,1h),5.19-4.95(m,1h),3.82-3.45(m,4h),3.01-2.96(m,2h),2.87-2.84(m,1h),2.19
‑ꢀ
2.08(m,5h),1.57-1.55(m,3h).
[0423]
lcms:[m 1]=454.2。
[0424]
手性hplc的条件:
[0425]
流动相:0.1%二乙胺 70%正己烷 30%异丙胺
[0426]
流速:0.4ml/min
[0427]
运行时间:30min
[0428]
柱温:30℃
[0429]
检测波长:254nm。
[0430]
经手性hplc分析对比,该手性合成方法得到的手性化合物41-p2的保留时间(14.088min)与化合物41经sfc拆分得到的p2具有相同的保留时间(14.018min),因此确定41-p2的绝对构型如式所示。
[0431]
生物测试例
[0432]
1.vanin-1重组酶活性抑制实验
[0433]
精确称取一定质量的化合物,用dmso以及反应缓冲液(50mmtrisbase,50mmkcl,1.6mm半胱胺,0.005%brij35,ph8.0,现配现用)配置化合物,使化合物的最高浓度为10000nm,按4倍梯度稀释,配制成10个不同浓度的化合物工作液;
[0434]
对于重组人vanin-1(百奥莱博,jn0618)活性抑制反应,先将2.5μl的化合物工作液和5μl重组人vanin-1蛋白混合,室温孵育15min后,加入2.5μl的pantetheine7-amino-4-trifluoromethylcoumarin底物,使10μl反应体系中,重组人vanin-1的终浓度为62.5pm,底物pantetheine7-amino-4-trifluoromethylcoumarin的终浓度为45μm,反应在384孔板(perkinelmer,6007280)中进行,dmso的终浓度为1%在酶标仪上设定激发光为405nm、发射光为505nm,25℃动力学读数1小时。收集第30分钟的原始数据进行数据处理和分析,再用graphpadprism8软件拟合浓度-效应曲线,并计算化合物浓度的ic
50
。数据如表-1所示。
[0435]
2.人血浆vanin-1酶活性抑制实验
[0436]
对于人血浆活性抑制反应,先将2.5μl的化合物工作液和5μl稀释100倍后的人血浆混合,室温孵育15min后,加入2.5μl的pantetheine7-amino-4-trifluoromethylcoumarin(内部合成)底物,使10μl反应体系中,底物pantetheine7-amino-4-trifluoromethylcoumarin的终浓度为25μm,反应在384孔板(perkinelmer,6007280)中进行,dmso的终浓度为1%。在酶标仪上设定激发光为405nm、发射光为505nm,25℃动力学读数1小时。收集第30分钟的原始数据进行数据处理和分析,再用graphpadprism8软件拟合浓度-效应曲线,并计算化合物浓度的ic
50
。数据如表-1和表-2所示。
[0437]
表-1
[0438]
实施例vanin-1 ic
50
(nm)实施例vanin-1 ic
50
(nm)化合物113化合物231.2化合物2411化合物2430.8化合物3323化合物251.3化合物41化合物2614.5化合物565化合物270.4化合物66化合物280.8化合物746化合物290.4化合物818化合物302.1化合物936化合物326.8化合物108化合物331.1化合物1140化合物3410.7化合物1217化合物350.5
化合物1374化合物3623.4化合物1438化合物370.4化合物150.4化合物380.1化合物16211化合物391.8化合物1782化合物410.2化合物181.4化合物41-p20.3化合物202.3化合物a11.25化合物2239.1
ꢀꢀ
[0439]
表-2
[0440][0441][0442]
化合物a为参考cn109476645a中实施例142的方法制备得到。
[0443]
3.药物-药物相互作用(cyp酶抑制活性ic
50
测试)
[0444]
本实验采用鸡尾酒法进行待测化合物对cyp酶的1a2,2b6,2c8,2c19,2c9,2d6和 3a4亚型酶活性抑制研究,以测得待测化合物对以上几种cyp酶亚型活性抑制的ic50 值。其中实验所用cyp酶探针底物为:phenacetin(1a2),bupropion(2b6),amodiaquine (2c8),mephenytoin(2c19),diclofenac(2c9),dextromethorphan(2d6)和testosterone (3a4/5)。微粒体在实验体系中的终浓度为0.1mg/ml。pbs buffer为50mm的k2hpo4 缓冲液。待测化合物浓度分别为50μm,12.5μm,3.125μm,0.781μm,0.195μm,0.0488m 合。将相应探针底物和微粒体加入pbs中,混合均匀,分装至每个反应体系中,随后分别在对应反应体系中加入对照化合物/待测化合物/dmso溶液,将反应体系混合均匀,37℃水浴预孵育5min,加入10mm nadph溶液并混合均匀,置于37℃水浴反应10min,反应结束后,加入内标乙腈溶液终止反应。4000rpm离心,取上清溶液加入等体积的纯水混合均匀,lc-ms/ms(ab triple quard 5500)分别分析各探针底物反应生成物的量,并利用测得的产物生成量用graphpad prism 5进行ic
50
的计算和图表制作。所测部分发明化合物数据如表-3所示。
[0445]
表-3
[0446][0447]
从表-3可知,化合物41-p2对所测cyp酶基本无抑制作用。
[0448]
4.体外肝微粒体稳定性实验
[0449]
本实验微粒体在实验体系中的终浓度为0.5mg/ml。pbs buffer为50mm的k2hpo4 缓冲液。待测化合物浓度为1μm。将微粒体加入pbs中,混合均匀,分装至每个反应体系中,分别在对应的反应体系中加入对照化合物/待测化合物,混合均匀后,置于37℃水浴预孵育5min,加入20mm的nadph溶液,混合均匀,在37℃水浴条件下,启动反应。分别在反应时间点0min,10min,30min,60min和90min时,每个反应体系各取出 30μl的反应样品,立刻加入300μl的内标乙腈溶液终止反应。4000rpm离心后,取上清溶液加入等体积的纯水混合均匀,lc-ms/ms(ab triple quard 5500)分别检测每个时间点样品中化合物量,并用microsoft excel 2010计算出t1/2,in vitro clint(μl/min/mgprotein),clhepatic(ml/min/kg)等参数。所测部分发明化合物数据如表-4所示。
[0450]
表-4
[0451][0452]
从表-4可知,化合物41-p2的肝微粒体具有显著的稳定性。
[0453]
5.小鼠体内药代动力学评价
[0454]
实验使用溶媒为:dmso:solutol:pbs=5%:25%:70%(v/v/v)。配制方法:准确称量所需化合物,按比例加入一定体积的dmso,涡旋混匀完全溶解后,按上述比例依次加入solutol和pbs,混匀即可。实验中静脉(iv)给药组和口服(po)给药组所使用的溶媒为相同溶媒。静脉剂量为2mpk,口服剂量为5mpk。实验采血时间点:iv组: 0.083,0.25,0.5,1,2,4,7,24h。po组:0.25,0.5,1,2,4,7,24h每个时间点颈静脉采集全血200ul,edta-k2抗凝,立即在4000rpm*5min,4℃条件下离心,取上清,样品冻存于-80℃冰箱。血浆样品的处理:经含内标的acn/meoh(1:1,v/v)沉淀剂沉淀后,14000rpm离心5min,取上清进lc-ms/ms(ab triple quard 5500)分析,获得血药浓度,并通过winnolin 8.1版本的非房室模型进行参数计算。结果见表-5:
[0455]
表-5
[0456][0457]
注:表格中
“‑”
为不适用。
[0458]
从表-5可知,在所给剂量下化合物41-p2暴露量为10656.5hr*ng/ml,在小鼠体内表现出较高的生物利用度为86%,显示了本发明化合物具有优越的小鼠药代动力学性质。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。