1.本发明属于化学药物分析领域,具体涉及一种取代丁烯酰胺原料及制剂产品的杂质及其分析方法。
背景技术:
2.一种取代丁烯酰胺,为(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺化合物或其药学上可接受的盐、溶剂合物,临床主要用于肿瘤的治疗。
[0003]
在所述取代丁烯酰胺合成及储存过程中,因自身结构的特点,可能会产生明显的降解产物,包括自身的顺式异构体的产生,及合成过程中的一系列杂质。例如其中杂质a的结构式如下所示。
[0004]
化合物a
[0005]
现有的规定对高毒或基因毒性杂质的规定不充足。规范地进行杂质的研究,并将其控制在一个安全、合理的限度范围之内,将直接关系到上市药品的质量及安全性。
技术实现要素:
[0006]
发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供取代丁烯酰胺原料及制剂产品的杂质。
[0007]
本发明还要解决的技术问题为提供上述取代丁烯酰胺原料及制剂产品的杂质在质量检测方法中的用途。
[0008]
本发明另要解决的技术问题为提供上述取代丁烯酰胺原料及制剂产品的杂质的分析方法。
[0009]
本发明进一步要解决的技术问题是提供上述取代丁烯酰胺杂质化合物的制备方法。
[0010]
其中,所述取代丁烯酰胺为(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺化合物或其药学上可接受的盐、溶剂合物、或其药学上可接受的盐的溶剂合物,特别是其马来酸盐一水合物。
[0011]
为了解决上述第一个技术问题,本发明公开了式(i)所示化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂合物或前药,或其代谢物;
[0012][0013]
其中,-r为-nh2,-nhcoch3和-nhcocooh中的任意一种。
[0014]
其中,当-r为-nh2时,其具体的结构如式ii所示(化合物b);当-r为-nhcoch3时,其具体的结构如式iii所示(化合物c);当-r为-nhcocooh时,其具体的结构如式ⅳ所示(化合物d);
[0015][0016]
为了解决上述第二个技术问题,本发明公开了式(i)所示化合物,即化合物b或化合物c或化合物d在(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺化合物或其药学上可接受的盐、溶剂合物、或其药学上可接受的盐的溶剂合物的原料及其制剂产品质量检测方法中的用途;
[0017][0018][0019]
其中,-r为-nh2,-nhcoch3和-nhcocooh中的任意一种。
[0020]
其中,当-r为-nh2时,其具体的结构如式ii所示(化合物b);当-r为-nhcoch3时,其具体的结构如式iii所示(化合物c);当-r为-nhcocooh时,其具体的结构如式ⅳ所示(化合物d);
[0021][0022]
在一些实施方案中,式(i)所示化合物,即化合物b或化合物c或化合物d在(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐或马来酸盐水合物的原料及其制剂产品质量检测方法中的用途也在本发明的保护范围之内。
[0023]
在一些典型实施方案中,式(i)所示化合物,即化合物b或化合物c或化合物d在(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐或马来酸盐一水合物的原料及其制剂产品质量检测方法中的用途也在本发明的保护范围之内
[0024]
为了解决上述第三个技术问题,本发明公开了一种(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺化合物或其药学上可接受的盐、溶剂合物、或其药学上可接受的盐的溶剂合物的原料及其制剂产品质量检测方法,检测(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺化合物或其药学上可接受的盐、溶剂合物的原料及其制剂中式(i)所示化合物的含量,即化合物b或化合物c或化合物d的含量;
[0025][0026]
其中,-r为-nh2,-nhcoch3和-nhcocooh中的任意一种。
[0027]
其中,当-r为-nh2时,其具体的结构如式ii所示(化合物b);当-r为-nhcoch3时,其具体的结构如式iii所示(化合物c);当-r为-nhcocooh时,其具体的结构如式ⅳ所示(化合物d);
[0028][0029]
在一些实施方案中,所述的(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐或马来酸盐水合物的原料及其制剂产品质量检测方法为检测(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐或马来酸盐水合物原料及其制剂中式(i)所示化合物的含量,即化合物b或化合物c或化合物d的含量。
[0030]
在一些典型实施方案中,所述的(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐或马来酸盐一水合物的原料及其制剂产品质量检测方法为检测(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐或马来酸盐一水合物原料及其制剂中式(i)所示化合物的含量,即化合物b或化合物c或化合物d的含量。
[0031]
在一些实施方案中,用hplc检测化合物b的含量,通过该方法,杂质与取代丁烯酰
胺的分离度大于1.5。
[0032]
在一些实施方案中,所述hplc所采用的条件为:
[0033]
色谱柱:c18柱
[0034]
流动相:流动相a为ph 6.0-8.0的盐水溶液;流动相b为有机溶剂
[0035]
洗脱条件:流动相a:流动相b(v/v)=55-70:30-45
[0036]
流速:0.5-1.5ml/min
[0037]
柱温:15-40℃
[0038]
检测波长:255-265nm。
[0039]
在一些实施方案中,所述的流动相b为乙腈或甲醇;在一些典型的实施方案中,所述的流动相b为乙腈。
[0040]
在一些实施方案中,所述hplc所采用的条件为:
[0041]
色谱柱:c18柱
[0042]
流动相:流动相a为ph 6.0-8.0的盐水溶液;流动相b为有机溶剂
[0043]
洗脱条件:梯度洗脱
[0044]
流速:0.8-1.2ml/min
[0045]
柱温:15-40℃
[0046]
检测波长:255-265nm。
[0047]
在一些实施方案中,所述的流动相b为乙腈或甲醇;在一些典型的实施方案中,所述的流动相b为乙腈。
[0048]
在一些实施方案中,所述的梯度洗脱条件为,起始流动相a与流动相b的体积比为35-45:65-55,中间流动相a与流动相b的体积比为60-75:40-25,最终流动相a与流动相b的体积比为35-45:65-55。
[0049]
在一些典型实施方案中,在0-25min时,流动相a与流动相b的体积比为35-45:65-55,在25~50min时,流动相a与流动相b的体积比为60-75:40-25,在50min以后,流动相a与流动相b的体积比为35-45:65-55。
[0050]
在一些实施方案中,所述的梯度洗脱条件,见表1;
[0051]
表1流动相梯度洗脱参数
[0052]
时间(min)流动相a(%)流动相b(%)0396125396150703050.01396160停止 。
[0053]
在一些实施方案中,所述盐水溶液为乙酸盐水溶液、磷酸盐水溶液、钾盐水溶液、钠盐水溶液和铵盐水溶液中的任意一种;在一些典型实施方案中,所述盐水溶液为磷酸钾水溶液、磷酸二氢钾水溶液、磷酸氢二钾水溶液、磷酸钠水溶液、磷酸二氢钠水溶液、磷酸氢二钾水溶液和乙酸铵水溶液中的任意一种。
[0054]
在一些实施方案中,调节盐水溶液的ph为7.0。
[0055]
在一些实施方案中,所述盐水溶液的浓度为0.01-3wt%;在一些典型实施方案中,所述盐水溶液的浓度为0.01-2wt%;在一些更典型实施方案中,所述盐水溶液的浓度为0.1-2wt%。
[0056]
在一些实施方案中,所述盐水溶液为0.1-2wt%乙酸铵水溶液;在一些典型实施方案中,所述盐水溶液为ph 6.0-8.0的乙酸铵水溶液。
[0057]
在一些实施方案中,所述的柱温为20-30℃;在一些典型实施方案中,所述的柱温为25℃。
[0058]
在一些实施方案中,所述的流速为0.8-1.2ml/min;在一些典型实施方案中,所述的流速为1.0ml/min。
[0059]
在一些实施方案中,检测器为紫外检测器,所述的检测波长为258-264nm;在一些典型实施方案中,所述的检测波长为261nm。
[0060]
在一些实施方案中,检测方法中进样体积为10-100μl;在一些典型实施方案中,检测方法中进样体积为10μl。
[0061]
在一些实施方案中,配置对照品溶液或供试品溶液所用的溶剂为极性溶剂;在一些典型实施方案中,所述的溶剂为水和极性有机溶剂中的任意一种或两种组合;在一些更典型实施方案中,所述的溶剂为水和乙腈中的任意一种或两种组合;在一些最典型实施方案中,所述的溶剂为水和乙腈的组合。
[0062]
在一些实施方案中,水和极性溶剂的体积比为10-60:90-40;在一些典型实施方案中,水和极性溶剂的体积比为20-50:80-50;在一些更典型实施方案中,水和极性溶剂的体积比为30-50:70-50;在一些最典型实施方案中,水和极性溶剂的体积比为40:60。
[0063]
在一些实施方案中,c18色谱柱的直径为4.6mm,柱长为150mm或250mm,填料粒径为3.5μm或5μm;在一些典型实施方案中,c18色谱柱的直径为4.6mm,柱长为150mm或250mm,填料粒径为5μm;在一些更典型实施方案中,c18色谱柱的直径为4.6mm,柱长为250mm,填料粒径为5μm。
[0064]
在一些最典型实施方案中,液相色谱条件为用十八烷基硅烷键合硅胶为填充剂(agilent zorbax extend c18,4.6
×
250mm 5μm或效能相当的色谱柱),以1wt%乙酸铵水溶液(用三乙胺或乙酸调ph至7.0)为流动相a,乙腈为流动相b;按表1梯度洗脱;流速为每分钟1ml;检测波长为261nm;柱温为25℃;进样量10μl。
[0065]
在一些实施方案中,采用极性溶剂将取代丁烯酰胺配制成0.1-1mg/ml的溶液;在一些典型实施方案中,采用极性溶剂将取代丁烯酰胺配制成0.5mg/ml的溶液。
[0066]
在一些实施方案中,采用极性溶剂将式(i)所示的化合物配置成0.1-50μg/ml的溶液。
[0067]
为了解决上述第四个技术问题,本发明公开了式(i)所示化合物的制备方法;
[0068]
[0069]
其中,-r为-nh2,-nhcoch3和-nhcocooh中的任意一种。
[0070]
在一些实施方案中,当-r为-nh2,-nhcoch3时,即化合物b和化合物c的制备方法已公开于中发明专利cn202010683653.x“一种egfr类分子靶向抗肿瘤药物的制备方法”中。
[0071]
在一些实施方案中,当-r为-nhcocooh,即化合物d为(n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]乙二酸单酰胺)的制备方法为n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺与草酰氯单甲酯及三乙胺在有机溶剂下反应得到,或根据已公开于中发明专利cn202010683653.x“一种egfr类分子靶向抗肿瘤药物的制备方法”中原料药的制备方法制备得到。
[0072]
在一些实施方案中,化合物d的合成过程中,三乙胺和n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺先混合再加入草酰氯单甲酯。
[0073]
在一些实施方案中,化合物d的合成过程中,草酰氯单甲酯和n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺先混合再加入三乙胺。
[0074]
在一些实施方案中,n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺与草酰氯单甲酯和三乙胺的质量比为10:4-7:3-8;在一些典型实施方案中,三者的质量比为10:5.18:4.9。
[0075]
在一些实施方案中,化合物d的合成过程中,反应温度为0-30℃;在一些典型的实施方案中,化合物d的合成过程中,反应温度为10-20℃。
[0076]
在一些实施方案中,化合物d的合成过程中,选用其他可替代的纯化方法。
[0077]
有益效果:与现有技术相比,本发明具有如下优势:
[0078]
采用本发明的方法可以有效的检测取代丁烯酰胺原料药及制剂中的各有关物质,异构体、降解杂质及其他杂质与取代丁烯酰胺均可以有效分离,其杂质的分离度与取代丁烯酰胺大于1.5。利用本发明的方法可以快速准确地进行取代丁烯酰胺原料药及制剂的有关物质定量分析,保证取代丁烯酰胺原料药及制剂的质量可控。
附图说明
[0079]
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
[0080]
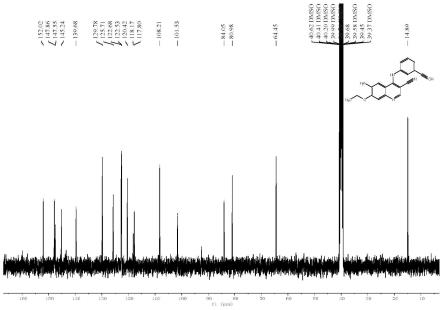
图1为化合物b的核磁碳谱图。
[0081]
图2为化合物b的核磁氢谱图。
[0082]
图3为化合物c的质谱谱图。
[0083]
图4为化合物d的核磁氢谱图。
[0084]
图5为取代丁烯酰胺及化合物a、b、c、d的液相色谱图。
[0085]
图6为系统适用性溶液在实施例5条件下检测的液相色谱图。
[0086]
图7为系统适用性溶液在实施例6条件下检测的液相色谱图。
具体实施方式
[0087]
以下结合实施例用于进一步描述本发明,但这些实施例并非限制着本发明的范围。取代丁烯酰胺、杂质化合物a、杂质化合物b、杂质化合物c、杂质化合物d均为公司自制;三乙胺来自阿拉丁,色谱级,若无特殊说明,其他试剂均可商业途径得到。
[0088][0089]
实施例1化合物b的合成
[0090]
50l反应釜中,加入1.155kg n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺,正丙醇18.48kg,搅拌均匀;加入0.52kg间氨基苯乙炔(间氨基苯乙炔与n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺摩尔比为:1.1:1);升温到90℃-95℃,反应3小时达到终点,降温到0℃-5℃,离心,固体用正丙醇洗涤,干燥,得到化合物b为1.544kg,为盐酸盐形式,收率96%,hplc测试含量为99.58%。化合物b的核磁如图1和图2所示,1h-nmr(d
6-dmso):9.19(s,1h),8.40(s,1h),7.26-7.30(m,1h),7.23(s,1h),7.19(s,1h),7.02-7.09(m,3h),5.51(s,1h),4.22-4.27(q,2h,j=6.8),4.14(s,1h),3.17(t,3h,j=6.8);
13
c-nmr(d
6-dmso):152.02,147.86,147.55,145.24,139.68,129.78,125.71,122.68,122.53,120.42,118.17,117.80,108.21,101.53,84.05,80.98,64.45,39.68,39.45,14.89。
[0091]
实施例2化合物c的合成
[0092]
50l反应釜中,加入1.00kgn-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺,正丙醇15.00kg,搅拌均匀;加入0.325kg间氨基苯乙炔(间氨基苯乙炔与n-(4-氯-3-氰基-7-乙氧基喹啉-6-基)乙酰胺摩尔比为:0.8:1);升温到沸腾回流,反应2.7小时达到终点,降温到0℃-10℃,离心,固体用正丙醇洗涤,干燥,得到化合物c为1.336kg,收率95%,含量为99.53%。化合物c的质谱如图3所示,m h
为371.2。结构确定:1h-nmr(d
6-dmso):9.11(s,1h),8.76(s,1h),8.14(s,1h),7.13-7.23(m,2h),6.98(s,3h),4.19-4.26(q,2h,j=6.9),4.05(s,1h),3.41-3.48(q,1h,j=6.9),2.11(s,3h),1.42-1.46(t,3h,j=6.9)。
[0093]
实施例3化合物d的合成
[0094]
将4.9g三乙胺,10g化合物b,5.18g草酰氯单甲酯,200ml四氢呋喃加入三口烧瓶中,在0℃下反应1h,后置于室温反应1h,倒入冰水,抽滤,得到的固体分散到1.5l乙醇中,加热搅拌,冷却抽滤,得到的固体溶于250ml甲醇中,室温下加入80ml的naoh水溶液(1mol/l),抽滤,再加入hcl调至ph为1,析出固体,抽滤,得到化合物d,收率为95%,纯度为97%。化合物d的核磁如图4所示,1h-nmr(d
6-dmso):9.97(s,1h),8.99(s,1h),8.64(s,1h),7.49(s,1h),7.36-7.40(t,1h,j=8.0),7.24-7.29(m,3h),4.33-4.39(q,2h,j=6.8),4.2(s,1h),1.45-1.49(t,2h,j=6.8)。
[0095]
实施例4:化合物a、化合物b、化合物c及化合物d作为标准品的应用
[0096]
仪器:agilent 1260高效液相色谱仪
[0097]
色谱柱:agilent zorbax extend(4.6mm
×
250mm,5μm)
[0098]
流动相:1wt%乙酸铵水溶液(用三乙胺或乙酸调ph至7.0)为流动相a,乙腈为流动相b;梯度洗脱程序如表1所示。
[0099]
检测波长:261nm
[0100]
流速:1.0ml/min
[0101]
柱温:25℃
[0102]
进样量:10μl
[0103]
稀释剂:乙腈-水(v/v 60:40)
[0104]
空白溶液即为稀释剂。
[0105]
各杂质的储备液:精密称取化合物a、化合物b、化合物c、化合物d适量,置棕色量瓶中,加乙腈-水(60:40v/v)超声溶解并稀释制成每1ml中约各含50μg的溶液作为杂质化合物a、杂质化合物b、杂质化合物c、杂质化合物d储备液。
[0106]
系统适用性溶液:精密称取(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐一水合物对照品约50mg,加乙腈-水(60:40)适量溶解后,分别加入上述杂质储备液,用乙腈-水(60:40)稀释,摇匀,制备成每1ml中约含取代丁烯酰胺为0.5mg、杂质化合物a为2μg、杂质化合物b为0.001mg、杂质化合物c为0.0025mg、杂质化合物d为0.001mg的溶液,作为系统适用性溶液。
[0107]
供试品溶液:精密称取(e)-n-[4-(3-乙炔基苯基)氨基-3-氰基-7-乙氧基喹啉-6-基]-4-(二甲基氨基)丁-2-烯酰胺马来酸盐一水合物(通过发明专利cn202010683653.x“一种egfr类分子靶向抗肿瘤药物的制备方法”中原料药的制备方法制备得到)12.5mg,置25ml棕色量瓶中,用乙腈-水(60:40)稀释剂溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
[0108]
测定:取空白溶剂、系统适用性溶液、供试品溶液各10μl,分别注入高效液相色谱仪,记录色谱图。
[0109]
如图5所示(图中的1为杂质化合物b,2为杂质化合物c,3为原料主峰,4为杂质化合物a,5为杂质化合物d),各杂质均有效分离,分离度均大于1.5。右手第1个峰为化合物b,保留时间在17分钟左右;右手第2个峰为化合物c,保留时间约为16分钟左右;主峰左侧第一个峰化合物a,保留时间在6分钟左右;左右第2个峰为化合物d,保留时间在3分钟左右。供试品溶液若出现待测的杂质,均可有效进行控制。
[0110]
实施例5:其他色谱条件
[0111]
色谱柱:agilent zorbax extend-c18(4.6
×
150mm,5μm)
[0112]
检测波长:261nm,流速:1.0ml/min,进样量:10μl,柱温:35℃
[0113]
流动相a:1wt%乙酸铵溶液(ph7.6)
[0114]
流动相b:乙腈
[0115]
梯度洗脱,如下:
[0116]
表2
[0117]
时间(min)流动相a(%)流动相b(%)08020206832405545503565553565568020618020
[0118]
溶液配制方法参照实施案例1。系统适用性溶液的色谱图见图6。结果表明,色谱柱温35℃、流动相a的ph为7.6,采用如表2所示的梯度洗脱时,杂质化合物b和化合物c无法分
离,分离度小于1.5。该色谱条件不能用于检测取代丁烯酰胺有关物质。
[0119]
实施例6:其他色谱条件
[0120]
色谱柱:agilent zorbax extend-c18(4.6
×
150mm,5μm)
[0121]
检测波长:261nm,流速:1.0ml/min,进样量:10μl,柱温:32℃
[0122]
流动相a:1wt%乙酸铵溶液(ph6.0)
[0123]
流动相b:乙腈
[0124]
梯度洗脱,如表2。
[0125]
溶液配制方法参照实施案例1。系统适用性溶液的色谱图见图7。结果表明,缓冲盐溶液ph6.0、柱温32℃,采用如表2所示的梯度洗脱时,主峰与后邻杂质出峰顺序变化,且分离较差,杂质化合物b和杂质化合物c分离度小于1.5,该色谱条件不能用于检测取代丁烯酰胺有关物质。
[0126]
本发明提供了一种采用高效液相色谱同时检测取代丁烯酰胺及其有关物质的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。