使用分子条形码进行准确碱基判定的方法
交叉引用
1.本技术要求于2019年6月12日提交的美国临时专利申请第62/860,462号的权益,其通过引用以其全部内容并入本文。
背景技术:
2.阐明整个人类基因组这一目标引起了对用于小规模和大规模应用的快速核酸(例如,脱氧核糖核酸(dna)或核糖核酸(rna))测序技术的兴趣。随着对人类疾病遗传基础知识的增加,高通量dna测序已被用于各种各样的临床应用。尽管核酸测序方法和系统在广泛的分子生物学和诊断应用中普遍存在,但此类方法和系统在准确的碱基判定(base calling)方面可能会遇到挑战。特别地,基于指示核苷酸掺入的量化特征性信号执行碱基判定的测序方法可能具有测序错误,其源于基本随机误差(例如,检测中的泊松噪声和来自生物化学过程的二项式噪声)和/或信号水平的不可预测的系统性变化,以及对于每个序列都可能不同的邻近序列(context)依赖性信号。此类信号变化和邻近序列依赖性信号可能导致序列判定方面的问题。
技术实现要素:
3.本文认识到需要改进的对序列的碱基判定。本文提供的方法和系统可以显著减少或消除由基本随机误差(例如,检测中的泊松噪声和来自生物化学过程的二项式噪声)导致的序列的碱基判定和/或同聚物长度估计中的误差,其通常可以通过重复次数的平方根来减少。本公开内容的方法和系统可以使用分子条形码将测序信号分组,聚集组内的测序信号,并将聚集的测序信号组合以生成共有序列。这样的方法和系统可以以非常低的单拷贝错误率实现对序列的准确和有效的碱基判定,这是使检测稀有事件的灵敏度最大化同时使特异性最大化(例如,使错误检测最小化)所需要的。
4.在一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对所述多个条形码化核酸分子进行测序以生成多个测序信号,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取(sequencing read);(c)使用对应于所述多个条形码序列的所述信号将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号包括对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的条形码序列不同的条形码序列的信号;(d)处理所述给定组内的所述测序信号以生成一个或多个聚集信号集,其中所述一个或多个聚集信号集不是测序读取;以及(e)将所述一个或多个聚集信号集组合以生成共有序列。
5.在一些实施方案中,在(e)中,所述组合包括执行碱基判定以鉴别单个碱基。在一些实施方案中,所述碱基判定通过对所述一个或多个聚集信号集中的每一个内的聚集信号彼此进行处理以生成所述共有序列来执行。在一些实施方案中,所述方法还包括将所述一
个或多个聚集信号集中的每一个内的所述聚集信号彼此平均以生成所述共有序列。在一些实施方案中,所述方法还包括对照参考处理所述共有序列以鉴别一个或多个遗传变体。在一些实施方案中,所述碱基判定通过对照参考信号对所述一个或多个聚集信号集中的每一个内的聚集信号进行处理以生成所述共有序列来执行。在一些实施方案中,所述多个核酸分子从受试者的身体样品获得。在一些实施方案中,所述多个核酸分子包括脱氧核糖核酸(dna)分子。在一些实施方案中,所述dna分子包括甲基化dna分子。在一些实施方案中,所述多个核酸分子包括核糖核酸(rna)分子。在一些实施方案中,在(a)中,所述条形码化包括将所述条形码分子与所述多个核酸分子连接。在一些实施方案中,所述多个条形码化核酸分子被非独特地条形码化。在一些实施方案中,所述多个条形码分子包括至少约100,000个不同的条形码。在一些实施方案中,所述多个条形码分子包含至少2个核苷酸取代的汉明距离。在一些实施方案中,所述多个测序信号包括模拟信号(analog signal)。在一些实施方案中,所述方法还包括在(c)之前或之后预处理所述多个测序信号以去除系统误差。在一些实施方案中,所述方法还包括在(b)之前扩增所述多个条形码化核酸分子。在一些实施方案中,所述扩增包括聚合酶链反应(pcr)。在一些实施方案中,所述扩增包括重组酶聚合酶扩增(rpa)。在一些实施方案中,所述多个测序信号通过大规模平行阵列测序生成。在一些实施方案中,所述多个测序信号通过流式测序生成。在一些实施方案中,(c)和(d)与(b)的所述测序实时或接近实时执行。在一些实施方案中,(e)与(b)的所述测序实时或接近实时执行。
6.在一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,所述多个测序信号通过使用多个条形码分子对所述多个核酸分子进行条形码化并对所述多个条形码化核酸分子进行测序来生成,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;以及可操作地耦合到所述数据库的一个或多个计算机处理器,其中所述一个或多个计算机处理器被单独地或共同地编程用于:使用对应于所述多个条形码序列的所述信号将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号包括对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的条形码序列不同的条形码序列的信号;处理所述给定组内的所述测序信号以生成一个或多个聚集信号集,其中所述一个或多个聚集信号集不是测序读取;以及将所述一个或多个聚集信号集组合以生成共有序列。
7.在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对所述多个条形码化核酸分子进行测序以生成多个测序信号,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;(c)处理对应于所述多个条形码序列的所述信号以鉴别所述多个测序信号中的每一个的所述条形码序列;(d)使用所述鉴别的条形码序列将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的鉴别的条形码序列不同的鉴别的条形码序列;(e)处理所述给定组内的所述测序信号以生成一个或多个聚集信号集,其中所述一个或多个聚集信号集不是测序读取;以及(f)将所述一个或多个聚集信号集
组合以生成共有序列。
8.在一些实施方案中,在(f)中,所述组合包括执行碱基判定以鉴别单个碱基。在一些实施方案中,所述碱基判定通过对所述一个或多个聚集信号集中的每一个内的聚集信号彼此进行处理以生成所述共有序列来执行。在一些实施方案中,所述处理包括将所述一个或多个聚集信号集中的每一个内的所述聚集信号彼此平均以生成所述共有序列。在一些实施方案中,所述方法还包括对照参考处理所述共有序列以鉴别一个或多个遗传变体。在一些实施方案中,所述碱基判定通过对照参考信号对所述一个或多个聚集信号集中的每一个内的聚集信号进行处理以生成所述共有序列来执行。在一些实施方案中,所述多个核酸分子从受试者的身体样品获得。在一些实施方案中,所述多个核酸分子包括脱氧核糖核酸(dna)分子。在一些实施方案中,所述dna分子包括甲基化dna分子。在一些实施方案中,所述多个核酸分子包括核糖核酸(rna)分子。在一些实施方案中,在(a)中,所述条形码化包括将所述条形码分子与所述多个核酸分子连接。在一些实施方案中,所述多个条形码化核酸分子被非独特地条形码化。在一些实施方案中,所述多个条形码分子包括至少约10万个不同的条形码。在一些实施方案中,所述多个条形码分子包含至少2个核苷酸取代的汉明距离。在一些实施方案中,所述多个测序信号包括模拟信号。在一些实施方案中,所述方法还包括在(d)之前或之后预处理所述多个测序信号以去除系统误差。在一些实施方案中,所述方法还包括在(b)之前扩增所述多个条形码化核酸分子。在一些实施方案中,所述扩增包括聚合酶链反应(pcr)。在一些实施方案中,所述扩增包括重组酶聚合酶扩增(rpa)。在一些实施方案中,所述多个测序信号通过大规模平行阵列测序生成。在一些实施方案中,所述多个测序信号通过流式测序生成。在一些实施方案中,(d)和(e)与(b)的所述测序实时或接近实时执行。在一些实施方案中,(f)与(b)的所述测序实时或接近实时执行。
9.在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,所述多个测序信号通过使用多个条形码分子对所述多个核酸分子进行条形码化并对所述多个条形码化核酸分子进行测序来生成,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;以及可操作地耦合到所述数据库的一个或多个计算机处理器,其中所述一个或多个计算机处理器被单独地或共同地编程用于:处理对应于所述多个条形码序列的所述信号以鉴别所述多个测序信号中的每一个的所述条形码序列;使用所述鉴别的条形码序列将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的鉴别的条形码序列不同的鉴别的条形码序列;处理所述给定组内的所述测序信号以生成一个或多个聚集信号集,其中所述一个或多个聚集信号集不是测序读取;以及将所述一个或多个聚集信号集组合以生成共有序列。
10.在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对所述多个条形码化核酸分子进行测序以生成多个测序信号,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;(c)使用对应于所述多个条形码序列的所述信号将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号包括对应于所述多个条形码序
列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的条形码序列不同的条形码序列的信号;(d)处理所述给定组内的所述测序信号以生成一个或多个估计序列,其中所述一个或多个估计序列中的每一个包含多个估计碱基判定;以及(e)将所述一个或多个估计序列组合以生成共有序列。
11.在一些实施方案中,所述一个或多个估计序列包括多个估计序列,并且所述共有序列是基于所述多个估计序列中的多数投票生成的。在一些实施方案中,所述方法还包括对照参考处理所述共有序列以鉴别一个或多个遗传变体。在一些实施方案中,所述多个核酸分子从受试者的身体样品获得。在一些实施方案中,所述多个核酸分子包括脱氧核糖核酸(dna)分子。在一些实施方案中,所述dna分子包括甲基化dna分子。在一些实施方案中,所述多个核酸分子包括核糖核酸(rna)分子。在一些实施方案中,在(a)中,所述条形码化包括将所述条形码分子与所述多个核酸分子连接。在一些实施方案中,所述多个条形码化核酸分子被非独特地条形码化。在一些实施方案中,所述多个条形码分子包括至少约10万个不同的条形码。在一些实施方案中,所述多个条形码分子包含至少2个核苷酸取代的汉明距离。在一些实施方案中,所述多个测序信号包括模拟信号。在一些实施方案中,所述方法还包括在(c)之前或之后预处理所述多个测序信号以去除系统误差。在一些实施方案中,所述方法还包括在(b)之前扩增所述多个条形码化核酸分子。在一些实施方案中,所述扩增包括聚合酶链反应(pcr)。在一些实施方案中,所述扩增包括重组酶聚合酶扩增(rpa)。在一些实施方案中,所述多个测序信号通过大规模平行阵列测序生成。在一些实施方案中,所述多个测序信号通过流式测序生成。在一些实施方案中,(c)和(d)与(b)的所述测序实时或接近实时执行。在一些实施方案中,(e)与(b)的所述测序实时或接近实时执行。
12.在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,所述多个测序信号通过使用多个条形码分子对所述多个核酸分子进行条形码化并对所述多个条形码化核酸分子进行测序来生成,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;以及可操作地耦合到所述数据库的一个或多个计算机处理器,其中所述一个或多个计算机处理器被单独地或共同地编程用于:使用对应于所述多个条形码序列的所述信号将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号包括对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的条形码序列不同的条形码序列的信号;处理所述给定组内的所述测序信号以生成一个或多个估计序列,其中所述一个或多个估计序列中的每一个包含多个估计碱基判定;以及将所述一个或多个估计序列组合以生成共有序列。
13.在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对所述多个条形码化核酸分子进行测序以生成多个测序信号,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;(c)处理对应于所述多个条形码序列的所述信号以鉴别所述多个测序信号中的每一个的所述条形码序列;(d)使用所述鉴别的条形码序列将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的条形码序列不同的鉴别
的条形码序列;(e)处理所述给定组内的所述测序信号以生成一个或多个估计序列,其中所述一个或多个估计序列中的每一个包含多个估计碱基判定;以及(f)将所述一个或多个估计序列组合以生成共有序列。
14.在一些实施方案中,所述一个或多个估计序列包括多个估计序列,并且所述共有序列是基于所述多个估计序列中的多数投票生成的。在一些实施方案中,所述方法还包括对照参考处理所述共有序列以鉴别一个或多个遗传变体。在一些实施方案中,所述多个核酸分子从受试者的身体样品获得。在一些实施方案中,所述多个核酸分子包括脱氧核糖核酸(dna)分子。在一些实施方案中,所述dna分子包括甲基化dna分子。在一些实施方案中,所述多个核酸分子包括核糖核酸(rna)分子。在一些实施方案中,在(a)中,所述条形码化包括将所述条形码分子与所述多个核酸分子连接。在一些实施方案中,所述多个条形码化核酸分子被非独特地条形码化。在一些实施方案中,所述多个条形码分子包括至少约10万个不同的条形码。在一些实施方案中,所述多个条形码分子包含至少2个核苷酸取代的汉明距离。在一些实施方案中,所述多个测序信号包括模拟信号。在一些实施方案中,所述方法还包括在(d)之前或之后预处理所述多个测序信号以去除系统误差。在一些实施方案中,所述方法还包括在(b)之前扩增所述多个条形码化核酸分子。在一些实施方案中,所述扩增包括聚合酶链反应(pcr)。在一些实施方案中,所述扩增包括重组酶聚合酶扩增(rpa)。在一些实施方案中,所述多个测序信号通过大规模平行阵列测序生成。在一些实施方案中,所述多个测序信号通过流式测序生成。在一些实施方案中,(d)和(e)与(b)的所述测序实时或接近实时执行。在一些实施方案中,(f)与(b)的所述测序实时或接近实时执行。
15.在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,所述多个测序信号通过使用多个条形码分子对所述多个核酸分子进行条形码化并对所述多个条形码化核酸分子进行测序来生成,所述多个测序信号包括对应于所述多个条形码序列的信号,其中所述多个测序信号不是测序读取;以及可操作地耦合到所述数据库的一个或多个计算机处理器,其中所述一个或多个计算机处理器被单独地或共同地编程用于:处理对应于所述多个条形码序列的所述信号以鉴别所述多个测序信号中的每一个的所述条形码序列;使用所述鉴别的条形码序列将所述多个测序信号分组成多个组,其中所述多个组中的给定组的测序信号对应于所述多个条形码序列中(i)对于所述给定组是相同的并且(ii)与所述多个组中的其他组的鉴别的条形码序列不同的鉴别的条形码序列;处理所述给定组内的所述测序信号以生成一个或多个估计序列,其中所述一个或多个估计序列中的每一个包含多个估计碱基判定;以及将所述一个或多个估计序列组合以生成共有序列。
16.通过以下在其中仅示出和描述了本公开内容的说明性实施方案的详细描述,本公开内容的其他方面和优点对于本领域技术人员将变得明显。如将会认识到的,本公开内容能够具有其他和不同的实施方案,并且其若干细节能够在各个明显的方面进行修改,所有这些都不偏离本公开内容。因此,附图和说明书在本质上将被认为是说明性而非限制性的。援引并入
17.本说明书中提及的所有出版物、专利和专利申请均通过引用并入本文,其程度如同特别地且单独地指出每个单独的出版物、专利或专利申请通过引用而并入。在通过引用并入的出版物和专利或专利申请与本说明书中包含的公开内容相抵触的程度下,本说明书
旨在取代和/或优先于任何此类矛盾的材料。
附图说明
18.本发明的新颖特征在所附权利要求中具体阐述。通过参考以下对其中利用到本发明原理的说明性实施方案加以阐述的详细描述以及附图(本文也称为“图”),将会获得对本发明特征和优点的更好理解,在这些附图中:
19.图1示出了根据所公开的实施方案,图示使用分子条形码进行碱基判定的方法的流程图的示例。
20.图2示出了根据所公开的实施方案,多个扩增的条形码化文库片段信号读取的示例。
21.图3示出了根据所公开的实施方案,多个扩增的条形码化文库片段信号读取的示例,这些条形码化文库片段信号读取已基于其条形码被分类并分组成更小的条形码特异性池。
22.图4示出了根据所公开的实施方案,在每个条形码池内执行读取-读取比对的示例,其提供了可进行分析的模板拷贝组以提高信噪比(snr)和碱基判定准确度,从而允许基于单个输入拷贝的稀有变体判定。
23.图5示出了被编程或以其他方式配置用于实现本文提供的方法的计算机系统。
24.图6示出了使用tf1l模板的流信号和用于碱基判定的人类基因组训练神经网络模型生成的数据的示例。
25.图7示出了使用tf4l模板的流信号和用于碱基判定的人类基因组训练神经网络模型生成的数据的示例。
26.图8示出了使用tf3l模板的流信号和用于碱基判定的大肠杆菌基因组训练神经网络模型生成的数据的示例。
27.图9示出了使用tf4l模板的流信号和用于碱基判定的大肠杆菌基因组训练神经网络模型生成的数据的示例。
具体实施方式
28.虽然本文已经示出和描述了本发明的各种实施方案,但对于本领域技术人员明显的是,这样的实施方案仅以示例的方式提供。本领域技术人员可在不偏离本发明的情况下想到许多变化、改变和替代。应当理解,可以使用本文中所述的本发明的实施方案的各种替代方案。
29.如本文所用,术语“测序”通常是指用于生成或鉴别生物分子(诸如核酸分子)的序列的过程。这样的序列可以是核酸序列,其可以包括核酸碱基的序列。测序方法可以是大规模平行阵列测序(例如,illumina测序),其可使用固定在支持物如流动细胞或珠上的模板核酸分子来执行。测序方法可包括但不限于:高通量测序、下一代测序、边合成边测序、流式测序、大规模平行测序、鸟枪法测序、单分子测序、纳米孔测序、焦磷酸测序、半导体测序、连接测序、杂交测序、核糖核酸(rna)测序(rna-seq)(illumina)、数字基因表达(helicos)、单分子合成测序(smss)(helicos)、克隆单分子阵列(solexa)和maxim-gilbert测序。
30.如本文所用,术语“流式测序”通常是指边合成边测序(sbs)过程,在该过程中,循
环或非循环地引入的单核苷酸溶液产生被感测到(例如,通过检测来自dna延伸的荧光信号的检测器)的分离的脱氧核糖核酸(dna)延伸。
31.如本文所用的术语“受试者”通常是指具有正在进行处理或分析的生物样品的个体。受试者可以是动物或植物。受试者可以是哺乳动物,诸如人、狗、猫、马、猪或啮齿动物。受试者可患有或疑似患有疾病,诸如癌症(例如,乳腺癌、结直肠癌、脑癌、白血病、肺癌、皮肤癌、肝癌、胰腺癌、淋巴瘤、食管癌或宫颈癌)或感染性疾病。受试者可患有或疑似患有遗传病症,诸如软骨发育不全、α-1抗胰蛋白酶缺乏症、抗磷脂综合征、孤独症、常染色体显性多囊肾病、进行性神经性腓骨肌萎缩征(charcot-marie-tooth)、猫叫综合征、克罗恩病、囊性纤维化、痛性肥胖病(dercum disease)、唐氏综合征、杜安综合征(duane syndrome)、杜氏肌营养不良、莱顿第五因子血栓形成倾向、家族性高胆固醇血症、家族性地中海热、脆性x综合征、戈谢病、血色素沉着症、血友病、前脑无裂畸形、亨廷顿病、克林费尔特综合征、马方综合征、强直性肌营养不良、神经纤维瘤病、努南综合征、成骨不全、帕金森病、苯丙酮尿症、波伦异常、卟啉症、早衰、色素性视网膜炎、重度联合免疫缺陷、镰状细胞病、脊髓性肌萎缩、泰-萨克斯病(tay-sachs)、地中海贫血、三甲基胺尿症、特纳综合征、腭帆心脏面部综合征、wagr综合征或威尔逊病。
32.如本文所用,术语“样品”通常是指生物样品。生物样品的实例包括核酸分子、氨基酸、多肽、蛋白质、碳水化合物、脂肪或病毒。在一个实例中,生物样品是包括一个或多个核酸分子,诸如脱氧核糖核酸(dna)和/或核糖核酸(rna)的核酸样品。核酸分子可以是无细胞的或无细胞的核酸分子,诸如无细胞dna或无细胞rna。核酸分子可衍生自多种来源,包括人类、哺乳动物、非人哺乳动物、猿、猴、黑猩猩、爬行动物、两栖动物或鸟类来源。此外,可从含有无细胞序列的各种动物流体提取样品,该流体包括但不限于血液、血清、血浆、玻璃体、痰液、尿液、泪液、汗液、唾液、精液、粘膜排泄物、粘液、脊髓液、羊水、淋巴液等。无细胞多核苷酸可能是胎儿起源的(通过取自妊娠受试者的流体),也可能衍生自受试者自身的组织。
33.如本文所用,术语“核酸”或“多核苷酸”通常是指包含一个或多个核酸亚单位或核苷酸的分子。核酸可包含选自腺苷(a)、胞嘧啶(c)、鸟嘌呤(g)、胸腺嘧啶(t)和尿嘧啶(u)或其变体的一种或多种核苷酸。核苷酸通常包含核苷和至少1、2、3、4、5、6、7、8、9、10个或更多个磷酸(po3)基团。核苷酸可包含核碱基、五碳糖(核糖或脱氧核糖)以及一个或多个磷酸基团。
34.核糖核苷酸是其中的糖为核糖的核苷酸。脱氧核糖核酸是其中的糖为脱氧核糖的核苷酸。核苷酸可以是核苷单磷酸或核苷多磷酸。核苷酸可以是脱氧核糖核苷多磷酸,例如,脱氧核糖核苷三磷酸(dntp),其可选自脱氧腺苷三磷酸(datp)、脱氧胞苷三磷酸(dctp)、脱氧鸟苷三磷酸(dgtp)、尿苷三磷酸盐(dutp)和脱氧胸苷三磷酸(dttp)dntp,包含可检测的标签,诸如发光标签或标记(例如,荧光团)。核苷酸可包括任何可掺入生长的核酸链的亚单位。这样的亚单位可以是a、c、g、t或u,或者特定于一个或多个互补a、c、g、t或u,或者互补于嘌呤(即a或g,或其变体)或嘧啶(即c、t或u,或其变体)的任何其他亚单位。在一些实例中,核酸是脱氧核糖核酸(dna)、核糖核酸(rna)或其衍生物或变体。核酸可以是单链的或双链的。在一些情况下,核酸分子是环形的。
35.如本文所用,术语“核酸分子”、“核酸序列”、“核酸片段”、“寡核苷酸”和“多核苷酸”通常是指具有各种长度的多核苷酸,诸如脱氧核糖核酸或核糖核苷酸(rna)或其类似
物。核酸分子可具有至少约10个碱基、20个碱基、30个碱基、40个碱基、50个碱基、100个碱基、200个碱基、300个碱基、400个碱基、500个碱基、1千碱基(kb)、2kb、3kb、4kb、5kb、10kb、50kb或更大的长度。寡核苷酸通常由四种核苷酸碱基:腺嘌呤(a);胞嘧啶(c);鸟嘌呤(g);和胸腺嘧啶(t)(当多核苷酸为rna时,用尿嘧啶(u)替代胸腺嘧啶(t))的特定序列组成。因此,术语“寡核苷酸序列”是多核苷酸分子的字母表示;或者,该术语可适用于多核苷酸分子本身。这种字母表示可输入到具有中央处理单元的计算机中的数据库中,并用于生物信息学应用,如功能基因组学和同源性搜索。寡核苷酸可包含一个或多个非标准核苷酸、核苷酸类似物和/或修饰核苷酸。
36.如本文所用,术语“核苷酸类似物”可包括但不限于二氨基嘌呤、5-氟尿嘧啶、5-溴尿嘧啶、5-氯尿嘧啶、5-碘尿嘧啶、次黄嘌呤、黄嘌呤(xantine)、4-乙酰胞嘧啶、5-(羧基羟甲基)尿嘧啶、5-羧甲基氨基甲基-2-硫代尿苷、5-羧甲基氨基甲基尿嘧啶、二氢尿嘧啶、β-d-半乳糖基q核苷(beta-d-galactosylqueosine)、肌苷、n6-异戊烯基腺嘌呤、1-甲基鸟嘌呤、1-甲基肌苷、2,2-二甲基鸟嘌呤、2-甲基腺嘌呤、2-甲基鸟嘌呤、3-甲基胞嘧啶、5-甲基胞嘧啶、n6-腺嘌呤、7-甲基鸟嘌呤、5-甲基氨基甲基尿嘧啶、5-甲氧基氨基甲基-2-硫尿嘧啶、β-d-甘露糖基q核苷、5'-甲氧基羧甲基尿嘧啶、5-甲氧基尿嘧啶、2-甲基硫-d46-异戊烯基腺嘌呤、尿嘧啶-5-氧乙酸(v)、wybutoxosine、假尿嘧啶、q核苷(queosine)、2-硫胞嘧啶、5-甲基-2-硫尿嘧啶、2-硫尿嘧啶、4-硫尿嘧啶、5-甲基尿嘧啶、尿嘧啶-5-氧乙酸甲酯、尿嘧啶-5-氧乙酸(v)、5-甲基-2-硫尿嘧啶、3-(3-氨基-3-n-2-羧基丙基)尿嘧啶、(acp3)w、2,6-二氨基嘌呤、硒代磷酸(phosphoroselenoate)核酸等。在一些情况下,核苷酸可以包括在其磷酸部分的修饰,包括对三磷酸部分的修饰。另外,修饰的非限制性实例包括更大长度的磷酸链(例如,具有4、5、6、7、8、9、10或多于10个磷酸部分的磷酸链)、具有巯基部分的修饰(例如,α硫代三磷酸和β硫代三磷酸)或具有硒部分的修饰(例如,硒代磷酸核酸)。核酸分子还可在碱基部分(例如,在通常可用于与互补核苷酸形成氢键的一个或多个原子处和/或在通常不能与互补核苷酸形成氢键的一个或多个原子处)、糖部分或磷酸骨架处进行修饰。核酸分子还可含有胺修饰基团,诸如氨基烯丙基dutp(aa-dutp)和氨基己基丙烯酰胺dctp(aha-dctp),以允许胺反应性部分(诸如n-羟基琥珀酰亚胺酯(nhs))的共价连接。本公开的寡核苷酸中的标准dna碱基对或rna碱基对的替代物可提供更高的密度(单位为每立方毫米(mm)的比特数)、更高的安全性(例如,对天然毒素的意外或有意合成的抗性)、更容易的光程序化聚合酶辨别或更低的二级结构。核苷酸类似物可能够与用于核苷酸检测的可检测部分反应或结合。
37.如本文所用,术语“游离核苷酸类似物”通常是指未与另外的核苷酸或核苷酸类似物偶联的核苷酸类似物。游离核苷酸类似物可通过引物延伸反应掺入生长的核酸链中。
38.如本文所用,术语“引物”通常是指与模板核酸互补的多核苷酸。引物和模板核酸之间的互补性、同源性或序列同一性可能是有限的。引物的长度可以是8个核苷酸碱基至50个核苷酸碱基。引物的长度可大于或等于6个核苷酸碱基、7个核苷酸碱基、8个核苷酸碱基、9个核苷酸碱基、10个核苷酸碱基、11个核苷酸碱基、12个核苷酸碱基、13个核苷酸碱基、14个核苷酸碱基、15个核苷酸碱基、16个核苷酸碱基、17个核苷酸碱基、18个核苷酸碱基、19个核苷酸碱基、20个核苷酸碱基、21个核苷酸碱基、22个核苷酸碱基、23个核苷酸碱基、24个核苷酸碱基、25个核苷酸碱基、26个核苷酸碱基、27个核苷酸碱基、28个核苷酸碱基、29个核苷
酸碱基、30个核苷酸碱基、31个核苷酸碱基、32个核苷酸碱基、33个核苷酸碱基、34个核苷酸碱基、35个核苷酸碱基、37个核苷酸碱基、40个核苷酸碱基、42个核苷酸碱基、45个核苷酸碱基、47个核苷酸碱基或50个核苷酸碱基。
39.引物可表现出与模板核酸的序列同一性或同源性或互补性。引物与模板核酸之间的同源性或序列同一性或互补性可基于引物的长度。例如,若引物长度为约20个核酸,则其可包含10个或更多个与模板核酸互补的连续核酸碱基。
40.如本文所用,术语“引物延伸反应”通常是指引物与模板核酸链的结合,然后是引物的延伸。其还可包括双链核酸的变性以及引物链与变性的模板核酸链中的一个或两个的结合,然后是引物的延伸。引物延伸反应可用于通过使用酶(聚合酶)以模板指导的方式将核苷酸或核苷酸类似物掺入引物。
41.如本文所用的术语“聚合酶”通常指能够催化聚合反应的任何酶。聚合酶的实例包括但不限于核酸聚合酶。聚合酶可以是天然存在的或是合成的。在一些情况下,聚合酶具有相对较高的持续合成能力。示例性的聚合酶是φ29聚合酶或其衍生物。聚合酶可以是聚合作用的酶。在一些情况下,使用转录酶或连接酶(即催化键形成的酶)。聚合酶的实例包括dna聚合酶、rna聚合酶、热稳定聚合酶、野生型聚合酶、修饰聚合酶、大肠杆菌(e.coli)dna聚合酶i、t7 dna聚合酶、噬菌体t4 dna聚合酶φ29(phi29)dna聚合酶、taq聚合酶、tth聚合酶、tli聚合酶、pfu聚合酶、pwo聚合酶、vent聚合酶、deepvent聚合酶、ex-taq聚合酶、la-taq聚合酶、sso聚合酶、poc聚合酶、pab聚合酶、mth聚合酶、es4聚合酶、tru聚合酶、tac聚合酶、tne聚合酶、tma聚合酶、tea聚合酶、tih聚合酶、tfi聚合酶、platinum taq聚合酶、tbr聚合酶、tfl聚合酶、pfutubo聚合酶、pyrobest聚合酶、pwo聚合酶、kod聚合酶、bst聚合酶、sac聚合酶、klenow片段、具有3'至5'外切核酸酶活性的聚合酶及其变体、修饰产物和衍生物。在一些情况下,聚合酶是单亚单位聚合酶。聚合酶可具有高持续合成能力,即聚合酶在不释放核酸模板的情况下连续地将核苷酸掺入核酸模板的能力。在一些情况下,聚合酶是经修饰以接受双脱氧核苷酸三磷酸的聚合酶,例如具有667y突变的taq聚合酶(参见例如,tabor等人,pnas,1995,92,6339-6343,其为了所有目的通过引用整体并入本文)。在一些情况下,聚合酶是具有修饰的核苷酸结合的聚合酶,其可能对核酸测序有用,非限制性实例包括thermosequenas聚合酶(ge life sciences)、amplitaq fs(thermofisher)聚合酶和sequencing pol聚合酶(jena bioscience)。在一些情况下,聚合酶被基因工程化为对双脱氧核苷酸具有辨别性,例如测序酶dna聚合酶(thermofisher)。
42.如本文所用的术语“支撑物”通常是指固体支撑物,诸如载玻片、珠子、树脂、芯片、阵列、基质、膜、纳米孔或凝胶。固体支撑物可以是例如平面基板(诸如玻璃、塑料、硅等)上的珠子或基板的孔内的珠子。基板可具有表面性质,诸如纹理、图案、微结构涂层、表面活性剂或其任何组合,以将珠子保持在期望位置(诸如待与检测器可操作通信的位置)。基于珠的支持物的检测器可以被配置为保持基本相同的读取速率,而与珠的大小无关。所述支撑物可以是流动池或开放基板。此外,所述支撑物可包括生物支撑物、非生物支撑物、有机支撑物、无机支撑物或其任何组合。支持物可与检测器进行光通信、可与检测器物理接触、可与检测器相隔一定距离或其任何组合。支持物可具有多个可独立寻址的位置。核酸分子可在多个可独立寻址位置的给定可独立寻址位置处固定至支持物。多个核酸分子中的每一个与支持物的固定可借助于衔接物的使用。支持物可与检测器光学耦合。在支持物上的固定
可借助于衔接物。
43.如本文所用,术语“标记”通常是指能够与诸如核苷酸类似物等物种偶联的部分。在一些情况下,标记可以是发射可被检测的信号(或减少已发射的信号)的可检测标记。在一些情况下,这样的信号可指示一个或多个核苷酸或核苷酸类似物的掺入。在一些情况下,标记可与核苷酸或核苷酸类似物偶联,其中核苷酸或核苷酸类似物可用于引物延伸反应。在一些情况下,标记可在引物延伸反应后与核苷酸类似物偶联。在一些情况下,标记可与核苷酸或核苷酸类似物特异性反应。偶联可以是共价的或非共价的(例如,通过离子相互作用、范德华力等)。在一些情况下,可经由接头偶联,该接头可以是可切割的,诸如光可切割(例如,在紫外光下可切割)、化学可切割(例如,经由还原剂,诸如二硫苏糖醇(dtt)、三(2-羧基乙基)膦(tcep))或酶可切割(例如,经由酯酶、脂肪酶、肽酶或蛋白酶)。
44.在一些情况下,标记可以是光学活性的。在一些实施方案中,光学活性标记是光学活性染料(例如,荧光染料)。染料的非限制性实例包括sybr绿、sybr蓝、dapi、碘化丙锭、hoeste、sybr金、溴化乙锭、吖啶、原黄素、吖啶橙、吖啶黄素、荧光香豆素(fluorcoumanin)、椭圆玫瑰树碱、道诺霉素、氯喹、偏端霉素d、色霉素、乙菲啶(homidium)、光神霉素、多吡啶钌、氨茴霉素、菲啶和吖啶、溴化乙锭、碘化丙锭、碘化己锭、二氢乙锭、乙锭同型二聚体-1和乙锭同型二聚体-2、单叠氮化乙锭和acma、hoechst 33258、hoechst 33342、hoechst 34580、dapi、吖啶橙、7-aad、放线菌素d、lds751、羟脒(hydroxystilbamidine)、sytox blue、sytox green、sytox orange、popo-1、popo-3、yoyo-1、yoyo-3、toto-1、toto-3、jojo-1、lolo-1、bobo-1、bobo-3、po-pro-1、po-pro-3、bo-pro-1、bo-pro-3、to-pro-1、to-pro-3、to-pro-5、jo-pro-1、lo-pro-1、yo-pro-1、yo-pro-3、picogreen、oligreen、ribogreen、sybr gold、sybr green i、sybr green ii、sybr dx、syto-40、syto-41、syto-42、syto-43、syto-44、syto-45(蓝)、syto-13、syto-16、syto-24、syto-21、syto-23、syto-12、syto-11、syto-20、syto-22、syto-15、syto-14、syto-25(绿)、syto-81、syto-80、syto-82、syto-83、syto-84、syto-85(橙)、syto-64、syto-17、syto-59、syto-61、syto-62、syto-60、syto-63(红)、荧光素、异硫氰酸荧光素(fitc)、四甲基异硫氰酸罗丹明(tritc)、罗丹明、四甲基罗丹明、r-藻红蛋白、cy-2、cy-3、cy-3.5、cy-5、cy5.5、cy-7、德克萨斯红(texas red)、phar-red、别藻蓝蛋白(apc)、sybr green i、sybr green ii、sybr gold、celltracker green、7-aad、乙锭同型二聚体i、乙锭同型二聚体ii、乙锭同型二聚体iii、溴化乙锭、伞形酮、曙红、绿色荧光蛋白、赤藓红、香豆素、甲基香豆素、芘、孔雀绿、茋、萤光黄、级联蓝(cascade blue)、二氯三嗪胺荧光素、丹磺酰氯、荧光镧系络合物(如包含铕和铽的那些络合物)、羧基四氯荧光素、5-羧基荧光素和/或6-羧基荧光素(fam)、vic、5-碘乙酰胺基荧光素或6-碘乙酰胺基荧光素、5-{[2-5-(乙酰基巯基)-琥珀酰基]氨基}荧光素和5-{[3-5-(乙酰基巯基)-琥珀酰基]氨基}荧光素(samsa-荧光素)、丽丝胺罗丹明b磺酰氯、5-羧基罗丹明和/或6-羧基罗丹明(rox)、7-氨基-甲基-香豆素、7-氨基-4-甲基香豆素-3-乙酸(amca)、bodipy荧光团、8-甲氧基芘-1,3,6-三磺酸三钠盐、3,6-二磺酸-4-氨基-萘二甲酰亚胺、藻胆蛋白、alexafluor 350、alexafluor 405、alexafluor 430、alexafluor 488、alexafluor 532、alexafluor 546、alexafluor 555、alexafluor 568、alexafluor 594、alexafluor 610、alexafluor 633、alexafluor 635、alexafluor 647、alexafluor 660、alexafluor 680、alexafluor 700、alexafluor 750和alexafluor 790染料、dylight 350、dylight 405、
dylight 488、dylight550、dylight 594、dylight 633、dylight 650、dylight 680、dylight755和dylight 800染料,或者其他荧光团。
[0045]
在一些实例中,标记可以是核酸嵌入剂染料。实例包括但不限于溴化乙锭、yoyo-1、sybr绿和evagreen。能量供体与能量受体之间、嵌入剂与能量供体之间或者嵌入剂与能量受体之间的近场相互作用可导致独特信号的生成或信号幅度的改变。例如,这样的相互作用可导致猝灭(即导致非辐射能量衰减的从供体到受体的能量转移)或福斯特共振能量转移(即导致辐射能量衰减的从供体到受体的能量转移)。标记的其他实例包括电化学标记、静电标记、比色标记和质量标签。
[0046]
如本文所用的术语“猝灭剂”通常是指能够降低发射的信号的分子。标记可以是猝灭剂分子。例如,模板核酸分子可被设计成发射可检测信号。包含猝灭剂的核苷酸或核苷酸类似物的掺入可减少或消除信号,然后检测到该减少或消除。在一些情况下,如本文其他部分所述,在核苷酸或核苷酸类似物掺入后,可发生使用猝灭剂的标记。猝灭剂的实例包括black hole猝灭剂染料(biosearch technologies),诸如bh1-0、bhq-1、bhq-3、bhq-10;qsy染料荧光猝灭剂(来自molecular probes/invitrogen),诸如qsy7、qsy9、qsy21、qsy35和其他猝灭剂如dabcyl和dabsyl;cy5q和cy7q以及暗花菁染料(ge healthcare)。其信号可与上述猝灭剂一起减少或消除的供体分子的实例包括荧光团,诸如cy3b、cy3或cy5;dy猝灭剂(dyomics),诸如dyq-660和dyq-661;荧光素-5-马来酰亚胺;7-二乙基氨基-3-(4'-马来酰亚胺基苯基)-4-甲基香豆素(cpm);n-(7-二甲基氨基-4-甲基香豆素-3-基)马来酰亚胺(dacm)和atto荧光猝灭剂(atto-tec gmbh),诸如atto 540q、580q、612q、647n、atto-633-碘乙酰胺、四甲基罗丹明碘乙酰胺或atto-488碘乙酰胺。在一些情况下,标记可以是不自猝灭的类型,例如二胺(bimane)衍生物,诸如单溴二胺(monobromobimane)。
[0047]
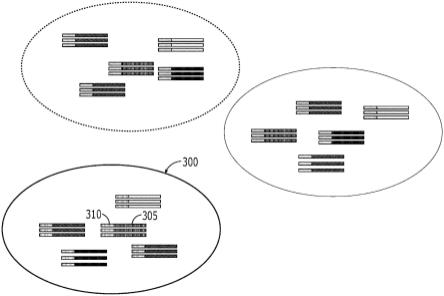
如本文所用,术语“检测器”通常是指能够检测信号的装置,该信号包括指示存在或不存在掺入的核苷酸或核苷酸类似物的信号。在一些情况下,检测器可包括可检测信号的光学和/或电子组件。术语“检测器”可用于检测方法中。检测方法的非限制性实例包括光学检测、光谱检测、静电检测、电化学检测等。光学检测方法包括但不限于荧光测定法和紫外-可见光吸收。光谱检测方法包括但不限于质谱、核磁共振(nmr)波谱和红外光谱。静电检测方法包括但不限于基于凝胶的技术,例如凝胶电泳。电化学检测方法包括但不限于在对扩增产物进行高效液相色谱分离后对扩增产物的电化学检测。
[0048]
如本文所用,术语“信号”、“信号序列”、“序列信号”和“测序信号”通常是指与dna分子或dna的克隆群体相关的一系列信号(例如,荧光测量值),包括原始数据。可使用高通量测序技术(例如,流式边合成边测序(sbs))获得此类信号。可对此类信号进行处理以获得估算序列(例如,在初步分析期间)。
[0049]
如本文所用,术语“序列”或“序列读取”通常是指在测序过程中进行的一系列核苷酸定位(assignment)(例如,通过碱基判定)。这样的序列可衍生自信号序列(例如,在初步分析期间)。序列读取可以是通过基于信号序列进行初步碱基判定来估计或估算的序列读取,然后可以对估计或估算的序列读取进行进一步的碱基判定分析或校正以产生最终序列读取(例如,使用本文公开的信噪比(snr)增强技术)。
[0050]
如本文所用的术语“同聚物”通常是指0,1,2,
…
,n个顺序的核苷酸的序列。例如,含有顺序的a核苷酸的同聚物可表示为a、aa、aaa,
…
,最多n个顺序的a核苷酸。
[0051]
如本文所用,术语“hpn截短”通常是指处理一个或多个序列的集合的方法,使得具有大于或等于整数n的长度的一个或多个序列的集合中的每个同聚物被截短为长度n的同聚物。例如,序列“agggggt”到3个碱基的hpn截短可导致“agggt”的截短序列。
[0052]
如本文所用,术语“类似物比对”通常是指将信号序列与参考信号序列进行比对。
[0053]
如本文所用,术语“邻近序列依赖性”或“邻近序列依赖关系”通常是指与局部序列、相对核苷酸表示或基因组位点的信号相关性。给定序列的信号可因邻近序列依赖性而变化,邻近序列依赖性可取决于局部序列、序列的相对核苷酸表示或序列的基因组位点。
[0054]
阐明整个人类基因组这一目标引起了对用于小规模和大规模应用的快速核酸(例如,dna)测序技术的兴趣。随着对人类疾病遗传基础知识的增加,高通量dna测序已被用于各种各样的临床应用。尽管核酸测序方法和系统在广泛的分子生物学和诊断应用中普遍存在,但此类方法和系统在准确的碱基判定方面可能会遇到挑战。特别地,基于指示核苷酸掺入的量化特征性信号执行碱基判定的测序方法可能具有测序错误,例如,其源于基本随机误差(例如,检测中的泊松噪声和来自生物化学过程的二项式噪声)和/或信号水平的不可预测的系统性变化,以及对于每个序列都可能不同的邻近序列依赖性信号。此类信号变化和邻近序列依赖性信号可能导致序列判定方面的问题。
[0055]
本文认识到需要至少解决上述问题的改进的对序列的碱基判定。本文提供的方法和系统可以显著减少或消除由基本随机误差(例如,检测中的泊松噪声和来自生物化学过程的二项式噪声)导致的序列的碱基判定和/或同聚物长度估计中的误差,其通常可以通过重复次数的平方根来减少。本公开内容的方法和系统可以使用分子条形码将测序信号分组,聚集组内的测序信号,并将聚集的测序信号组合以生成共有序列。这样的方法和系统可以以非常低的单拷贝错误率实现对序列的准确和有效的碱基判定和/或同聚物长度估计,这是使检测稀有事件(例如,序列或部分序列的稀有实例)的灵敏度最大化同时使特异性最大化(例如,使错误检测最小化)所需要的。
[0056]
流式边合成边测序(sbs)程序通常包括执行重复的dna延伸循环,其中核苷酸和/或标记的类似物的单个种类相继呈递给引物-模板-聚合酶复合物,然后在互补的情况下(与引物-模板-聚合酶复合物中的生长链互补)掺入该核苷酸。可针对模板的每个克隆群体(例如,珠或集落)测量每个流的产物。所得核苷酸掺入物可通过对应于零、一或更多个顺序的掺入物或者与之相关的明确区分性信号来检测和量化。在相同种类的核苷酸(例如,规范碱基类型)与生长链上(例如,在同聚物片段中)的连续位置互补的情况下,流可能导致多次掺入到生长链中。序列的准确的碱基判定和/或同聚物长度估计可以包括对这样的多个顺序的掺入物的量化,其可以包括对每个流中的集落上掺入的0,1,2,
…
,n个顺序的核苷酸的每种可能的情况的特征性信号进行量化。例如,顺序的a核苷酸的集可表示为a、aa、aaa,
…
,最多n个顺序的a核苷酸。
[0057]
在一些情况下,序列的准确的碱基判定和/或同聚物长度估计可能会由于基本随机误差(例如,检测中的泊松噪声和来自生物化学过程的二项式噪声,其通常可以通过重复次数的平方根来减少)和/或信号水平的不可预测的系统性变化而遇到挑战,其中任何一个都可能导致碱基判定误差。在一些情况下,仪器和检测系统可通过监视仪器诊断和大量集落之间的共模行为来校准和移除。序列的准确的碱基判定和/或同聚物长度估计还可能由于对于每个序列可能不同的邻近序列依赖性信号而遇到挑战。例如,在稀释标记的核苷酸
的荧光测量的情况下,邻近序列既可影响标记类似物的数目(用于并入标记类似物的可变耐受性),也可影响单个标记类似物的荧光(例如,受
±
5碱基的局部邻近序列影响的染料的量子产率,如[kretschy等人,sequence-dependent fluorescence of cy3-and cy5-labeled double-stranded dna,bioconjugate chem.,27(3),第840-848页]所述,其通过引用整体并入本文)。实际上,通过染料终止剂桑格循环测序,已经鉴别出对于3碱基邻近序列的信号的实质性系统变化(例如,如[zakeri等人,peak height pattern in dichloro-rhodamine and energy transfer dye terminator sequencing,biotechniques,25(3),第406-10页]所述,其通过引用整体并入本文)。
[0058]
本公开内容提供了使用分子条形码通过对测序应用(例如,适用于流式sbs)进行条形码分组来有效增强模拟信号的改进的序列的碱基判定和/或同聚物长度估计的方法和系统。所述方法和系统可包括算法步骤,以从与核苷酸流相对应的给定的一系列序列信号准确且有效地确定碱基判定和/或同聚物长度。
[0059]
在各个方面,诸如单个序列信号的信噪比(snr)较差可能导致碱基准确性较差从而导致基因组比对不准确的情况下,本公开内容的方法和系统可应用于在最终碱基判定之前提高这样的序列信号的snr。这些方法和系统可以包括获得输入核酸分子的样品,将多个不同条形码中的条形码附接至单个输入核酸分子上以产生多个条形码化核酸分子,以及扩增该多个条形码化核酸分子以产生扩增子的文库。该文库可以包含初始多个条形码化核酸分子的精确拷贝片段(具有相同的条形码和序列),以及其等位基因拷贝和等位基因变体,其通常可以共享分子条形码和片段终点(例如,起点和终点)。本公开内容的方法和系统可以包括将精确拷贝片段分组在一起(例如,已经从相同的初始模板分子扩增),并且在组内聚集或组合它们的信号以显著提高序列信号的snr,从而实现更准确的碱基判定和/或同聚物长度估计。
[0060]
执行序列信号的这样的snr增强的一种方法可以包括将所有多个n个序列读取相互比较,并将最佳匹配分组在一起。然而,这样的方法在计算上可能是代价非常大的,因为该操作的计算复杂度可能是n2阶(以大o符号计),当n非常大时(例如,接近10亿个输入核酸样品片段,这是诸如人类全基因组测序的应用的标称量),其可能在计算上有问题。
[0061]
图1示出了根据所公开的实施方案,图示使用分子条形码进行碱基判定的方法100的流程图的示例。首先,可以对多个初始模板分子进行条形码化,并且可以生成初始模板分子的条形码和未知序列的信号(如在105中)。接下来,可以通过条形码化信号(例如,通过信号相关性)对初始模板分子的未知序列进行分类(如在110中),然后通过测序信号(例如,通过相关性)进行进一步子分组(如在115中)或基于未知序列的估计的碱基判定进行进一步子分组(如在120中)。备选地,可以基于条形码序列(例如,通过条形码信号的碱基判定生成)对初始模板分子的未知序列进行分类(如在125中),然后通过测序信号进行进一步子分组(如在130中)或基于未知序列的估计的碱基判定(如在135中)进行进一步子分组。最后,可以根据组合的信号(如在140中)进行未知序列的碱基判定或者根据来自估计序列的共有序列的碱基判定(如在145中)进行未知序列的碱基判定。
[0062]
如图2所示,本公开内容的方法和系统可以包括制备核酸分子200的输入样品,由此将核酸分子的输入样品的每个初始模板分子205与多个条形码210之一连接。在一些实施方案中,核酸分子200的输入样品的每个初始模板分子205与多个条形码210之一唯一地连
接,从而产生各自具有不同条形码的多个条形码化核酸分子(例如,使得多个条形码化核酸分子中的任何对附接或连接至不同的条形码)。
[0063]
在对多个初始模板分子进行条形码化之后,可以将多个条形码化核酸分子扩增至足够的程度(例如,扩增循环次数),使得存在合理的可能性(例如,至少约50%、至少约60%、至少约70%、至少约80%、至少约90%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%、至少约99.9%或至少约99.99%)为每个初始模板分子获得超过一个精确拷贝的平均数量(例如,扩增子的数量)。
[0064]
本公开内容的方法可以在不将全部多个估算序列读取中的估算序列读取彼此比对(例如,在全部多个估算序列读取中相互比对估算序列读取)的情况下执行,从而降低碱基判定和/或同聚物长度评估的计算复杂性。备选地,本公开内容的方法可以在不将全部多个序列信号中的序列信号彼此比对(例如,在全部多个序列信号中相互比对序列信号)的情况下执行,从而降低碱基判定和/或同聚物长度评估的计算复杂性。
[0065]
在一些实施方案中,可以将每个序列信号或估算序列读取根据其条形码信号(例如,对应于分子条形码的模拟信号或估算序列读取,该分子条形码附接至生成该估算序列读取的片段)分类或分组到不同的条形码池中(例如,条形码池300),如图3所示(每个片段包含对应于初始模板分子的较长输入序列305,以及对应于连接分子条形码310的较短条形码序列)。由于条形码池300可包含具有相同分子条形码310的序列信号或估算序列读取,在随后的分析中可将序列信号或估算序列读取解释或处理为可能来自核酸分子输入样品的相同初始模板分子。条形码池300内的序列信号或估算序列读取也可能对应于核酸分子输入样品的不同初始模板分子(例如,具有序列305和序列315)。可以基于模拟分类(例如,将具有相同分子条形码的模拟信号的序列信号分组在一起)或基于数字化条形码(例如,将具有相同分子条形码的估算序列读取分组在一起)进行分组。
[0066]
在一些实施方案中,考虑到输入样品的分子多样性,多个条形码可以包含足够数量的碱基,使得初始模板分子可被独特或非独特地标记和鉴别。多个条形码可以包含1个碱基、2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基、16个碱基、17个碱基、18个碱基、19个碱基、20个碱基或超过20个碱基。一般而言,多个n碱基条形码可足以对具有约4n个初始模板分子的样品进行独特地条形码化。
[0067]
在一些实施方案中,可将多个条形码设计成使得多个条形码中的任何一对条形码之间的编辑距离(例如,汉明距离)足以避免混淆(例如,由扩增、复制、测序、碱基判定和/或同聚物长度评估中的单个碱基或几个碱基错误引起),从而实现包含1个碱基、2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基、16个碱基、17个碱基、18个碱基、19个碱基、20个碱基或超过20个碱基的错误的错误检测和/或错误纠正。在一些实施方案中,可将多个条形码设计成使得条形码的碱基数目的子集用于错误检查或校正(ecc)目的(例如,类似于在数据通信中使用奇偶校验位)。
[0068]
如图4所示,在将条形码化文库片段的序列信号或估算序列读取分组成条形码组之后(例如,条形码池300),可以将每个条形码组内的序列信号或估算序列读取进行相互(例如,相关的)比较,并且可以鉴别相同的序列信号或估算序列读取并将其进一步分组(例
如,在条形码组内)成代表相同初始模板分子的家族(例如,具有相同条形码310的三个相同序列信号或估算序列读取305的家族)。在通过初始模板分子分组成家族之后,可以在每个家族内将比对的序列信号或估算序列读取组合以产生每个家族具有更高snr(例如,平均值)的单个序列信号。与具有较低snr的单个序列信号或估算序列读取相比,这种组合的序列信号或估算序列读取可以进行更准确的碱基判定、比对和更置信的遗传变体评估。因为这些单独的序列信号或估算序列读取源自单个初始模板分子,所以它们代表单个等位基因,从而显著地简化了分析。在一些实施方案中,该过程可以仅通过模拟信号处理步骤完成,直到碱基判定。
[0069]
作为计算效率的数字示例,假设处理用多个105个条形码进行条形码化的多个109个单独的估算序列读取。执行简单的读取-读取比对可能需要o(10
18
)阶数的相关操作。相比之下,可以执行本公开内容的方法处理相同的用多个105个条形码进行条形码化的多个109个单独的估算序列读取,通过执行109个条形码的分类操作,然后执行相关运算;从而实现计算量减少等于条形码文库多样性的因数(例如,在这种情况下,5个数量级或因数为10,000)。因此,本公开内容的方法可有利地用于基于初始模板核酸分子的少量或单个输入拷贝来执行稀有变体判定,从而由于模拟信号增强方法而实现碱基判定和/或同聚物长度评估的效率和准确性的显著增益。使用重复sbs对菌落进行有效的模拟信号增强
[0070]
在一些实施方案中,本公开内容的方法可以包括通过对簇进行边合成边测序(sbs)(或类似的)测序,随后进行合成拷贝的变性和第二测序过程来减少由化学和检测过程引起的随机信号变化。与第二sbs操作相关的检测和化学的随机变化可以是独立的,并且可以与第一信号平均以减少噪声。可以根据需要重复该过程以将随机误差减少到期望或目标水平。该方法的优点可以包括尽管扫描和sbs成本与上述并行拷贝方法成倍增加,但仅产生单个拷贝的制备和基底成本。
[0071]
在本公开内容的各个方面,用于对多个核酸分子进行测序的方法可以包括(i)通过序列信号或条形码序列进行分类,(ii)通过序列信号或条形码序列进行子分组,并将子组内的序列信号或条形码序列聚集。用于对多个核酸分子进行测序的方法可以包括使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子。接下来,该方法可以包括对多个条形码化核酸分子进行测序以生成多个测序信号。多个测序信号可以包括对应于多个条形码序列的信号,并且该多个测序信号可以不是测序读取。备选地,该方法可以包括对多个条形码化核酸分子进行测序以生成多个估算测序读取。
[0072]
接下来,该方法可以包括使用对应于多个条形码序列的信号将多个测序信号分组成多个组。多个组中的给定组的测序信号可以包括对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的条形码序列的信号。备选地,该方法可以包括使用对应于多个条形码序列的估算序列读取将多个估算序列读取分组成多个组。多个组中的给定组的估算序列读取可以包括多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的条形码序列。
[0073]
接下来,该方法可以包括处理给定组内的测序信号以生成一个或多个聚集信号
集。一个或多个聚集信号集可以不是测序读取。接下来,该方法可以包括将一个或多个聚集信号集组合以生成核酸分子的共有序列。备选地,该方法可以包括聚集给定组内的估算序列读取以生成一个或多个聚集序列读取集。通过由条形码信号进行分类并且通过测序信号进行子分组的碱基判定
[0074]
在一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对多个条形码化核酸分子进行测序以生成多个测序信号,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;(c)使用对应于多个条形码序列的信号将多个测序信号分组成多个组,其中多个组中的给定组的测序信号包括对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的条形码序列的信号;(d)处理给定组内的测序信号以生成一个或多个聚集信号集,其中该一个或多个聚集信号集不是测序读取;以及(e)将该一个或多个聚集信号集组合以生成共有序列。
[0075]
在一些实施方案中,(e)中的组合包括执行碱基判定以鉴别单个碱基。碱基判定可以通过对一个或多个聚集信号集中的每一个内的聚集信号彼此进行处理以生成共有序列来执行。在一些实施方案中,该方法还包括将一个或多个聚集信号集中的每一个内的聚集信号彼此平均以生成共有序列。可以将共有序列与参考进行比较以鉴别一个或多个遗传变体。
[0076]
在一些实施方案中,可包括dna(例如,甲基化dna)分子或rna分子的多个核酸分子从受试者的身体样品获得。条形码化可以包括将条形码分子与多个核酸分子连接。多个条形码化核酸分子可以被独特地或非独特地条形码化。在一些实施方案中,多个条形码分子包括至少约10个、至少约100个、至少约1,000个、至少约10,000个或至少约100,000个不同的条形码。在一些实施方案中,多个测序信号包括模拟信号。在一些实施方案中,该方法还包括预处理多个测序信号以去除系统误差。在一些实施方案中,该方法还包括在(b)之前扩增多个条形码化核酸分子(例如,通过pcr或rpa)。在一些实施方案中,步骤(c)、(d)和/或(e)与(b)的测序实时或接近实时执行。
[0077]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,该多个测序信号通过使用多个条形码分子对多个核酸分子进行条形码化并对多个条形码化核酸分子进行测序来生成,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;以及可操作地耦合到数据库的一个或多个计算机处理器,其中一个或多个计算机处理器被单独地或共同地编程用于:使用对应于多个条形码序列的信号将多个测序信号分组成多个组,其中多个组中的给定组的测序信号包括对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的条形码序列的信号;处理给定组内的测序信号以生成一个或多个聚集信号集,其中该一个或多个聚集信号集不是测序读取;以及将该一个或多个聚集信号集组合以生成共有序列。
[0078]
在一些实施方案中,可以将多个估算序列及其相关序列信号聚集以鉴别局部邻近序列。然后,多个估算序列及其相关序列信号可堆叠在一起,在一些情况下使用与参考基因组的比对,以鉴别和分组与相同基因组位置相关联的核苷酸碱基。多个估算序列及其相关
序列信号可通过将估算序列相互比较来堆叠在一起,以鉴别共同的局部邻近序列。或者,多个估算序列及其相关序列信号可通过与参考序列比对而堆叠在一起。例如,多个估算序列(及其相关序列信号)可与参考基因组(例如,人参考基因组,诸如hg19或hg38)比对。或者,多个序列信号(及其相关的估算序列)可与参考信号比对。堆叠的估算序列及其相关信号可使用任何数目的可能包含邻近序列依赖性的连续碱基堆叠在一起,诸如2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基、16个碱基、17个碱基、18个碱基、19个碱基、20个碱基或多于20个碱基。
[0079]
使用这些可根据其分子条形码和/或n碱基邻近序列(例如,位于估算序列附近的n个连续碱基的数量)聚集和分组的估算序列,可以构建和训练邻近序列模型(例如,通过聚集针对特定基因组邻近序列的数据以观察任何系统行为),以学习如何解释用于准确的碱基判定的信号。开发邻近序列模型可以包括基于邻近序列依赖性信号和估算序列之间的相关性,分析多个相关联的序列信号以发现系统行为,以及开发用于预测碱基判定的规则,如本文其他部分所述。这样的相关性或邻近序列依赖性可包括在给定序列或信号之前和/或之后的多个碱基(例如,2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基、16个碱基、17个碱基、18个碱基、19个碱基、20个碱基或多于20个碱基)。例如,若基于邻近序列依赖性,“a”出现在第一序列(例如,“tctcg”)之后,则可预期第一信号水平(例如,标称信号的0.7),并且若“a”出现在第二序列(例如,“aaacc”)之后,则可预期第二信号水平(例如,预期标称信号的1.3)。这样的邻近序列依赖性可聚集到训练模型中以优化例如来自估算序列和/或序列信号的碱基判定。
[0080]
例如,邻近序列模型可以基于通过对具有已知序列(例如,来自合成模板dna分子)的dna分子进行测序而获得的估算序列和相关信号的分析而建立和训练(例如,使用机器学习技术)。这样的邻近序列模型可包含对应于位点的n碱基部分的预期序列信号(例如,信号幅度)(例如,其中n是至少1个碱基、至少2个碱基、至少3个碱基、至少4个碱基、至少5个碱基、至少6个碱基、至少7个碱基、至少8个碱基、至少9个碱基或至少10个碱基)。备选地或附加地,邻近序列模型可包括或包含对应于位点的n碱基部分的序列信号的分布、中值、平均值、众数、标准差、分位数、四分位距或其他量化或统计量度(例如,信号幅度)。
[0081]
本公开内容的方法和系统可包括仅使用先验已知序列(例如,双链序列)的算法,或同时评估一系列流测量值以确定包含最有可能产生观察结果的序列的一系列碱基判定(例如,最大似然序列测定)的算法。该算法可解释可能发生并影响序列信号的任何标记-标记相互作用,例如,猝灭。该算法还可解释可能发生并影响序列信号的任何已知的位置依赖性信号和/或任何光漂白作用。例如,邻近序列依赖性可能受核苷酸的混合群体(例如,包含天然核苷酸和修饰核苷酸)的流式测序的影响。这样的核苷酸混合群体可能在流式测序过程中竞争通过聚合酶的掺入,从而产生不同的邻近序列依赖性序列信号。
[0082]
该算法可并入已知序列的训练数据,该已知序列包含与同聚物信号变化具有显著相关性的每个邻近序列的一个或多个复制。对于待应用该算法的每个不同的分离的化学变化,可以重复这样的并入。
[0083]
该算法可包含辅助输出,其可包括量化噪声(例如,泊松或二项式随机变化)的评
估或其他质量评估,包括同聚物长度的置信区间或误差评估。输出还可以包括对化学过程参数(例如,温度)的动态评估,以及最有可能解释观察结果的标记部分。
[0084]
经训练的邻近序列模型可随后由一种或多种训练算法(例如,机器学习算法)应用,以预测碱基判定(例如,通过对具有未知序列的dna分子进行测序获得的多个估算序列和相关信号的碱基判定和/或同聚物长度)。这样的预测可包括优化或校正多个估算序列的碱基判定。或者,这样的预测可包括从多个序列信号确定碱基判定。例如,可以对包含未知序列的第二集合的dna分子进行测序,从而生成第二多个序列信号和估算序列。接下来,可以生成第二集合的dna分子的碱基判定,例如,至少基于(i)与第二多个序列信号相关联的第二多个估算序列和/或序列信号,(ii)第二多个估算序列,(iii)预期信号的至少一部分,(iv)已知序列,或(v)其组合。在一些实施方案中,这样的预测可以实时执行(例如,在测量序列信号的同时)。例如,实时可以包括小于1秒、十分之一秒、百分之一秒、毫秒或更短的响应时间。实时可以包括相对于另一过程或操作(例如,测量序列信号)同时或基本上同时发生的过程或操作(例如,生成碱基判定)。本文所述的所有操作,如训练算法、预测和/或生成碱基判定以及其他操作(诸如本文其他地方所述的那些操作)可被配置为能够实时发生或执行。通过由条形码序列进行分类并且通过测序信号进行子分组的碱基判定
[0085]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对多个条形码化核酸分子进行测序以生成多个测序信号,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;(c)处理对应于多个条形码序列的信号以鉴别多个测序信号中的每一个的条形码序列;(d)使用鉴别的条形码序列将多个测序信号分组成多个组,其中多个组中的给定组的测序信号对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的鉴别的条形码序列;(e)处理给定组内的测序信号以生成一个或多个聚集信号集,其中该一个或多个聚集信号集不是测序读取;以及(f)将该一个或多个聚集信号集组合以生成共有序列。
[0086]
在一些实施方案中,在(f)中,组合包括执行碱基判定以鉴别单个碱基。碱基判定可以通过对一个或多个聚集信号集中的每一个内的聚集信号彼此进行处理以生成共有序列来执行。在一些实施方案中,该方法还包括将一个或多个聚集信号集中的每一个内的聚集信号彼此平均以生成共有序列。可以将共有序列与参考进行比较以鉴别一个或多个遗传变体。
[0087]
在一些实施方案中,可包括dna(例如,甲基化dna)分子或rna分子的多个核酸分子从受试者的身体样品获得。条形码化可以包括将条形码分子与多个核酸分子连接。多个条形码化核酸分子可以被独特地或非独特地条形码化。在一些实施方案中,多个条形码分子包括至少约10个、至少约100个、至少约1,000个、至少约10,000个或至少约100,000个不同的条形码。在一些实施方案中,多个测序信号包括模拟信号。在一些实施方案中,该方法还包括预处理多个测序信号以去除系统误差。在一些实施方案中,该方法还包括预处理多个测序信号以去除系统误差。在一些实施方案中,该方法还包括在(b)之前扩增多个条形码化核酸分子(例如,通过pcr或rpa)。在一些实施方案中,步骤(d)、(e)和/或(f)与(b)的测序实
时或接近实时执行。
[0088]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,该多个测序信号通过使用多个条形码分子对多个核酸分子进行条形码化并对多个条形码化核酸分子进行测序来生成,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;以及可操作地耦合到数据库的一个或多个计算机处理器,其中一个或多个计算机处理器被单独地或共同地编程用于:处理对应于多个条形码序列的信号以鉴别多个测序信号中的每一个的条形码序列;使用鉴别的条形码序列将多个测序信号分组成多个组,其中多个组中的给定组的测序信号对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的鉴别的条形码序列不同的鉴别的条形码序列;处理给定组内的测序信号以生成一个或多个聚集信号集,其中该一个或多个聚集信号集不是测序读取;以及将该一个或多个聚集信号集组合以生成共有序列。通过由条形码信号进行分类并且通过序列进行子分组的碱基判定
[0089]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对多个条形码化核酸分子进行测序以生成多个测序信号,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;(c)使用对应于多个条形码序列的信号将多个测序信号分组成多个组,其中多个组中的给定组的测序信号包括对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的条形码序列的信号;(d)处理给定组内的测序信号以生成一个或多个估计序列,其中该一个或多个估计序列中的每一个包含多个估计碱基判定;以及(e)将该一个或多个估计序列组合以生成共有序列。
[0090]
在一些实施方案中,一个或多个估计序列包括多个估计序列,并且共有序列是基于多个估计序列中的多数投票生成的。可以将共有序列与参考进行比较以鉴别一个或多个遗传变体。在一些实施方案中,可包括dna(例如,甲基化dna)分子或rna分子的多个核酸分子从受试者的身体样品获得。条形码化可以包括将条形码分子与多个核酸分子连接。多个条形码化核酸分子可以被独特地或非独特地条形码化。在一些实施方案中,多个条形码分子包括至少约10个、至少约100个、至少约1,000个、至少约10,000个或至少约100,000个不同的条形码。在一些实施方案中,多个测序信号包括模拟信号。在一些实施方案中,该方法还包括预处理多个测序信号以去除系统误差。在一些实施方案中,该方法还包括在(b)之前扩增多个条形码化核酸分子(例如,通过pcr或rpa)。在一些实施方案中,步骤(c)、(d)和/或(e)与(b)的测序实时或接近实时执行。
[0091]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,该多个测序信号通过使用多个条形码分子对多个核酸分子进行条形码化并对多个条形码化核酸分子进行测序来生成,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;以及可操作地耦合到数据库的一个或多个计算机处理器,其中一个或多个计算机处理器被单独地或共同地编程用于:使用对应于多个条形码序列的信号将多个测序信号分组成多个组,其中多个组中的给定组的测序信号包括对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他
组的条形码序列不同的条形码序列的信号;处理给定组内的测序信号以生成一个或多个估计序列,其中该一个或多个估计序列中的每一个包含多个估计碱基判定;以及将该一个或多个估计序列组合以生成共有序列。通过由条形码序列进行分类并且通过序列进行子分组的碱基判定
[0092]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的方法,包括:(a)使用多个条形码分子对来自生物样品的多个核酸分子进行条形码化,以生成包含多个条形码序列的多个条形码化核酸分子;(b)对多个条形码化核酸分子进行测序以生成多个测序信号,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;(c)处理对应于多个条形码序列的信号以鉴别多个测序信号中的每一个的条形码序列;(d)使用鉴别的条形码序列将多个测序信号分组成多个组,其中多个组中的给定组的测序信号对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的条形码序列不同的鉴别的条形码序列;(e)处理给定组内的测序信号以生成一个或多个估计序列,其中该一个或多个估计序列中的每一个包含多个估计碱基判定;以及(f)将该一个或多个估计序列组合以生成共有序列。
[0093]
在一些实施方案中,一个或多个估计序列包括多个估计序列,并且共有序列是基于多个估计序列中的多数投票生成的。在一些实施方案中,该方法还包括对照参考处理共有序列以鉴别一个或多个遗传变体。在一些实施方案中,可包括dna(例如,甲基化dna)分子或rna分子的多个核酸分子从受试者的身体样品获得。条形码化可以包括将条形码分子与多个核酸分子连接。多个条形码化核酸分子可以被独特地或非独特地条形码化。在一些实施方案中,多个条形码分子包括至少约10个、至少约100个、至少约1,000个、至少约10,000个或至少约100,000个不同的条形码。在一些实施方案中,多个测序信号包括模拟信号。在一些实施方案中,该方法还包括预处理多个测序信号以去除系统误差。在一些实施方案中,该方法还包括预处理多个测序信号以去除系统误差。在一些实施方案中,该方法还包括在(b)之前扩增多个条形码化核酸分子(例如,通过pcr或rpa)。在一些实施方案中,步骤(d)、(e)和/或(f)与(b)的测序实时或接近实时执行。
[0094]
在另一方面,本公开内容提供了一种用于对多个核酸分子进行测序的系统,包括:存储多个测序信号的数据库,该多个测序信号通过使用多个条形码分子对多个核酸分子进行条形码化并对多个条形码化核酸分子进行测序来生成,该多个测序信号包括对应于多个条形码序列的信号,其中该多个测序信号不是测序读取;以及可操作地耦合到数据库的一个或多个计算机处理器,其中一个或多个计算机处理器被单独地或共同地编程用于:处理对应于多个条形码序列的信号以鉴别多个测序信号中的每一个的条形码序列;使用鉴别的条形码序列将多个测序信号分组成多个组,其中多个组中的给定组的测序信号对应于多个条形码序列中(i)对于给定组是相同的并且(ii)与多个组中的其他组的鉴别的条形码序列不同的鉴别的条形码序列;处理给定组内的测序信号以生成一个或多个估计序列,其中该一个或多个估计序列中的每一个包含多个估计碱基判定;以及将该一个或多个估计序列组合以生成共有序列。用于同聚物判定的方法
[0095]
本公开内容的方法和系统可用于对包含同聚物的序列执行准确且有效的碱基判定。这样的碱基判定可以作为测序过程,诸如执行核酸分子(例如,dna分子)的下一代测序
(例如,边合成边测序或流式测序)的一部分来执行。这样的核酸分子可以获自或衍生自来自受试者的样品。这样的受试者可患有疾病或疑似患有疾病。本文所述的方法和系统可用于显著减少或消除量化同聚物长度中的误差以及与邻近序列依赖性相关联的误差。这样的方法和系统可以实现同聚物的准确有效的碱基判定、同聚物长度的量化以及序列信号中邻近序列依赖性的量化。
[0096]
本文提供的方法和系统可用于直接判定同聚物长度,每个读取具有高准确性。此外,本文提供的方法和系统可包括将含有不确定长度的同聚物的临时量化读取(例如,估算或估计序列)与参考进行比对。可以使用对同聚物长度误差施加低罚分的算法来执行这样的比对。使用多个比对读取的统计能力、同聚物长度和不确定性的评估(例如,置信区间或误差评估),本文提供的方法和系统可基于所有读取(例如,对于纯合位点)或簇读取的共有序列来确定同聚物长度。备选地或组合地,本文提供的方法和系统可对簇(例如,对于杂合位点)进行共有序列判定。
[0097]
本公开内容的方法可以包括处理多个序列信号。这样的方法可用于通过比对读取的共有序列(诸如通过与hpn截短的参考序列进行比对)来确定同聚物长度。该方法可包括对核酸样品进行测序以提供多个序列信号和估算序列。从这样的估算序列,可以鉴别至少n个碱基的同聚物序列(例如,包含含有相同碱基的多个连续核苷酸的同聚物的序列)。这些已鉴别的估算同聚物序列随后可被截短为长度为n的碱基的同聚物序列,以产生一个或多个hpn截短序列。长度n可以是任何数目的多个碱基,诸如2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基或多于15个碱基。作为截短的同聚物比对的实例,给定序列中所有鉴别的长度为n或更大的同聚物可以被截短为长度n的同聚物,然后与参考进行比对。
[0098]
在截短后,一个或多个hpn截短序列可与一个或多个截短参考进行比对。这样的截短参考可以是hpn截短的,从而包含截短为长度n的一个或多个同聚物序列。在一个或多个hpn截短序列的比对后,可以从与一个或多个hpn截短的参考进行比对的一个或多个hpn截短序列生成共有序列。这样的共有序列可以包括长度为n的同聚物序列。可以基于比对的hpn截短序列、与比对的hpn截短序列相关联的序列信号或其组合生成共有序列。
[0099]
在一些实施方案中,处理多个序列信号可包括计算同聚物序列的长度估计误差。长度估计误差可包括同聚物序列长度(同聚物长度)的置信区间。例如,估算长度为5个碱基的同聚物的长度估计误差可包括[3,7]或5个碱基
±
2个碱基的置信区间。长度估计误差可至少基于与hpn截短参考比对的一个或多个hpn截短序列的信号的分布或估算的同聚物长度来计算。
[0100]
在一些实施方案中,处理多个序列信号可以包括预处理多个序列信号以去除系统误差。这样的预处理可以在截短鉴别的估算同聚物序列并将hpn截短序列与一个或多个截短参考进行比对之前执行。可以执行预处理以解决信号水平中的随机和不可预测的系统变化,其可导致量化同聚物长度时的误差。在一些情况下,可通过监测大量集落之间仪器诊断和共模行为来校准和去除仪器和检测的系统变化。
[0101]
在一些实施方案中,处理多个序列信号可包括确定同聚物序列的长度。可通过确定出现在由与多个序列信号相关联的比对的hpn截短序列生成的共有序列中的顺序核苷酸的数目来执行该确定。该确定可至少基于同聚物序列或与同聚物序列相关联的序列信号的
聚类来执行。
[0102]
在一些实施方案中,多个序列信号通过对受试者的核酸进行测序而产生。hpn截短参考可包括受试者物种的hpn截短参考基因组(例如,hpn截短的人参考基因组)。在一些情况下,当生成共有序列时,计算或分类的长度的数目可能受到限制,这至少基于受试者物种的倍性。多个序列信号和/或估算序列可以通过任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。
[0103]
本公开内容的方法可以包括量化多个序列信号和估算序列的邻近序列依赖性。这样的方法可用于通过用测定对已知基因组进行广泛训练来量化同聚物长度。该方法可包括对脱氧核糖核酸(dna)分子进行测序,以提供多个序列信号和估算序列。在一些情况下,dna分子包含已知序列。从这样的估算序列,可以鉴别至少n个碱基的同聚物序列(例如,包含含有相同碱基的多个连续核苷酸的同聚物的序列)。这些已鉴别的估算同聚物序列随后可被截短为长度n的碱基的同聚物序列,以产生一个或多个hpn截短序列。长度n可以是任何数目的多个碱基,诸如2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基或多于15个碱基。在截短后,一个或多个hpn截短序列可与一个或多个截短参考进行比对。这样的截短参考可以是hpn截短的,从而包含截短为长度n的一个或多个同聚物序列。在一个或多个hpn截短序列进行比对后,可以量化关联序列信号的邻近序列依赖性。这样的量化可至少基于(i)与一个或多个hpn截短参考比对的一个或多个hpn截短序列和/或与hpn截短参考比对的一个或多个hpn截短序列相关联的序列信号,(ii)已知序列,或(iii)其组合。
[0104]
在一些实施方案中,量化多个序列信号和估算序列的邻近序列依赖性包括对包含未知序列的第二集合的dna分子进行测序,从而生成第二多个序列信号和估算序列。从这样的估算序列,可以鉴别至少n个碱基的第二同聚物序列(例如,含有包含相同碱基的多个连续核苷酸的同聚物的序列)。这些鉴别的估算第二同聚物序列随后可被截短为长度n的碱基的同聚物序列,以产生一个或多个第二hpn截短序列。长度n可以是任何数目的多个碱基,诸如2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基或多于15个碱基。在截短后,一个或多个第二hpn截短序列可以与一个或多个hpn截短参考进行比对。在一个或多个hpn截短序列进行比对后,可以确定第二多个dna分子的同聚物长度。这样的确定可以至少基于(i)与hpn截短参考比对的一个或多个hpn截短序列和/或与hpn截短参考比对的一个或多个hpn截短序列相关联的序列信号,(ii)量化的邻近序列依赖性,或(iii)其组合。
[0105]
在一些实施方案中,量化的邻近序列依赖性针对给定邻近序列进行分类。这样的给定邻近序列可以是n碱基邻近序列,其中“n”是大于或等于2的整数、大于或等于3的整数、大于或等于4的整数、大于或等于5的整数、大于或等于6的整数、大于或等于7的整数、大于或等于8的整数、大于或等于9的整数、大于或等于10的整数、大于或等于11的整数、大于或等于12的整数、大于或等于13的整数、大于或等于14的整数、大于或等于15的整数、大于或等于16的整数、大于或等于17的整数、大于或等于18的整数、大于或等于19的整数或者大于或等于20的整数。
[0106]
例如,量化的邻近序列依赖性可针对n碱基邻近序列进行分类,其中初始序列判定(例如,估算序列)按n碱基邻近序列(例如,“tgttca”)分组。然后使用按n碱基邻近序列分组
的估算序列的关联信号来建立系统邻近序列映射。例如,对邻近序列内估算序列的单个碱基和同聚物(例如,分别为“t”、“g”、“tt”、“c”和“a”)的代表性信号测量(信号水平)及其信号变化进行测量并记录为历史数据。历史数据可以单独地或共同地存储在一个或多个数据库中。数据库可以包含任何数据结构,诸如图表、表格、列表、数组、图、索引、散列数据库、一个或多个图形或者任何其他类型的结构。
[0107]
作为另一实例,量化的邻近序列依赖性可针对n碱基邻近序列进行分类,其中hpn截短序列按n碱基邻近序列(例如,“tgttca”)分组。然后使用按n碱基邻近序列分组的hpn截短序列的关联信号来建立系统邻近序列映射。例如,对邻近序列内hpn截短序列的单个碱基和同聚物(例如,分别为“t”、“g”、“tt”、“c”和“a”)的代表性信号测量(信号水平)及其信号变化进行测量并记录为历史数据(例如,在本文所述的系统的数据库中)。
[0108]
在一些实施方案中,生成了邻近序列图谱,其包括信号与序列中掺入的连续核苷酸的数目(例如,同聚物长度)之间的数学关系。这样的关系可以表示为邻近序列特异性映射(邻近序列图谱)。真实序列(包含长度为2至4的同聚物)和真实序列的关联邻近序列依赖性信号的比较可以表明,由于邻近序列依赖性,同聚物的信号测量(信号水平)与同聚物的长度之间并没有完美的线性关系。这种非线性关系可导致估算同聚物长度的误差,该误差可随后使用历史数据和背景图谱进行校正。单调邻近序列(例如,按照同聚物长度严格递增的信号)可用于将一系列信号中的每一个映射到校正的同聚物长度。邻近序列图谱可用于训练一种或多种算法(例如,机器学习算法),以将信号转换为预测序列和/或同聚物长度。例如,可以将在估算序列中找到的每个局部邻近序列与聚合数据库进行比较,以检索可应用于转换的规则。
[0109]
在一些实施方案中,dna分子衍生自核糖核酸(rna)分子。例如,可以通过对rna分子执行逆转录以生成互补dna(cdna)分子或其衍生物来生成dna分子。多个序列信号和/或估算序列可以通过任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。在一些实施方案中,量化邻近序列依赖性包括针对多个位点中的每一个建立信号幅度与同聚物长度之间的关系。这样的关系可以表示为邻近序列特异性映射(邻近序列图谱)。
[0110]
本公开内容的方法可以包括量化多个序列信号和估算序列的邻近序列依赖性。这样的方法可以包括对脱氧核糖核酸(dna)分子进行测序,以提供多个序列信号和估算序列。在一些情况下,dna分子包含已知序列。从这样的估算序列,可以鉴别至少n个碱基的同聚物序列(例如,包含含有相同碱基的多个连续核苷酸的同聚物的序列)。这些已鉴别的估算同聚物序列随后可被截短为长度n的碱基的同聚物序列,以产生一个或多个hpn截短序列。长度n可以是任何数目的多个碱基,诸如2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基或多于15个碱基。在截短后,一个或多个hpn截短序列可与一个或多个截短参考进行比对。这样的截短参考可以是hpn截短的,从而包含截短为长度为n的一个或多个同聚物序列。在一个或多个hpn截短序列进行比对后,可以确定该hpn截短参考中的多个位点中的每一个的预期信号。这样的预期信号可至少基于以下来确定:(i)与hpn截短参考比对的一个或多个hpn截短序列和/或与hpn截短参考比对的一个或多个hpn截短序列相关联的序列信号,(ii)该已知序列,或(iii)其组合。
[0111]
在一些实施方案中,量化多个序列信号和估算序列的邻近序列依赖性包括对包含未知序列的第二集合的dna分子进行测序,从而生成第二多个序列信号和估算序列。从这样的估算序列,可以鉴别至少n个碱基的第二同聚物序列(例如,含有包含相同碱基的多个连续核苷酸的同聚物的序列)。这些鉴别的估算第二同聚物序列随后可被截短为长度为n的碱基的同聚物序列,以产生一个或多个第二hpn截短序列。长度n可以是任何数目的多个碱基,诸如2个碱基、3个碱基、4个碱基、5个碱基、6个碱基、7个碱基、8个碱基、9个碱基、10个碱基、11个碱基、12个碱基、13个碱基、14个碱基、15个碱基或多于15个碱基。在截短后,一个或多个第二hpn截短序列可以与一个或多个hpn截短参考进行比对。在一个或多个hpn截短序列进行比对后,可以确定第二多个dna分子的同聚物长度。这样的确定可以至少基于(i)与hpn截短参考比对的一个或多个hpn截短序列和/或与hpn截短参考比对的一个或多个hpn截短序列相关联的序列信号,(ii)量化的邻近序列依赖性,或(iii)其组合。
[0112]
在一些实施方案中,dna分子衍生自核糖核酸(rna)分子。例如,可以通过对rna分子执行逆转录以生成互补dna(cdna)分子或其衍生物来生成dna分子。多个序列信号和/或估算序列可以通过任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。在一些实施方案中,量化邻近序列依赖性包括针对多个位点中的每一个建立信号幅度与同聚物长度之间的关系。这样的关系可以表示为邻近序列特异性映射(邻近序列图谱)。
[0113]
本公开内容的方法可以包括处理多个序列信号。这样的方法可用于通过并入二次分析数据来确定同聚物长度。该方法可包括对核酸样品进行测序以提供多个序列信号和估算序列。可对多个序列信号和估算序列进行处理以确定包含同聚物序列的一个或多个序列的集合。还可对多个序列信号和估算序列进行处理以鉴别同聚物序列的至少一部分的存在和/或估计长度。可使用一种或多种算法通过将信号转换为同聚物长度(例如,使用邻近序列图谱或其他邻近序列依赖性信息)来鉴别同聚物序列的存在和/或估计长度。同聚物序列的估计长度可使用二次分析数据进行优化。这样的二次分析数据可用于提供或增强邻近序列依赖性信息。多个序列信号和/或估算序列可以通过任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。用于类似物比对的方法
[0114]
本公开内容的方法可以包括处理多个序列信号,以通过信号与参考信号(例如,类似物参考信号)的比对来确定碱基判定。该方法可包括对核酸样品进行测序以提供多个序列信号。多个序列信号可以与参考信号(例如,类似物参考信号)进行比对。至少基于比对的序列信号,可以鉴别包含碱基序列的参考位点。可以从与参考信号比对的多个序列信号生成共有序列。共有序列可包含n个碱基的序列。可以至少基于所鉴别的参考位点、该参考位点的序列的长度和参考信号(例如,类似物参考信号)来执行生成。
[0115]
在一些实施方案中,用于处理多个序列信号的方法可包括计算序列的长度估计误差。长度估计误差可包括序列长度的置信区间。例如,估算长度为5个碱基的序列的长度估计误差可包括[3,7]或5个碱基
±
2个碱基的置信区间。长度估计误差可至少基于与参考信号比对的多个序列信号的信号或估算的序列长度的分布来计算。
[0116]
在一些实施方案中,处理多个序列信号可以包括预处理多个序列信号以去除系统误差。这样的预处理可以在将多个序列信号与参考信号进行比对之前执行。可以执行预处
理以解决信号水平中的随机和不可预测的系统变化,其可导致对序列进行碱基判定时的误差。在一些情况下,可通过监测大量集落之间仪器诊断和共模行为来校准和去除仪器和检测的系统变化。
[0117]
在一些实施方案中,多个序列信号通过对受试者的核酸进行测序而产生。在一些情况下,当生成共有序列时,计算或分类的长度的数目可能受到限制,这至少基于受试者物种的倍性。多个序列信号可以由任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。
[0118]
本公开内容的方法可以包括量化多个序列信号的邻近序列依赖性。该方法可包括对脱氧核糖核酸(dna)或核糖核酸(rna)分子进行测序以提供多个序列信号。dna或rna分子可包含已知序列。多个序列信号可以与参考信号(例如,类似物参考信号)进行比对。可以在与该参考信号比对的多个序列信号中对邻近序列依赖性进行量化。邻近序列依赖性的量化可以至少基于已知序列来执行。在一些实施方案中,该比对可以包括执行一种或多种模拟信号处理算法。
[0119]
在一些实施方案中,量化多个序列信号的邻近序列依赖性包括对包含未知序列的第二集合的dna分子进行测序,从而生成第二多个序列信号。第二多个序列信号可以与参考信号(例如,类似物参考信号)进行比对。在第二多个序列信号进行比对后,可以确定第二多个dna分子的碱基判定。这样的确定可以至少基于与该参考信号比对的多个序列信号、量化的邻近序列依赖性或其组合。
[0120]
在一些实施方案中,dna分子衍生自核糖核酸(rna)分子。例如,可以通过对rna分子执行逆转录以生成互补dna(cdna)分子或其衍生物来生成dna分子。多个序列信号和/或估算序列可以通过任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。在一些实施方案中,量化邻近序列依赖性包括针对多个位点中的每一个建立信号幅度与碱基判定和/或序列长度之间的关系。这样的关系可以表示为邻近序列特异性映射(邻近序列图谱)。
[0121]
本公开内容的方法可以包括量化多个序列信号的邻近序列依赖性。该方法可包括对脱氧核糖核酸(dna)或核糖核酸(rna)分子进行测序以提供多个序列信号。dna或rna分子可包含已知序列。多个序列信号可以与参考信号(例如,类似物参考信号)进行比对。在多个序列信号与参考信号进行比对之后,可以针对参考信号中的多个位点中的每一个来确定预期信号。该确定可至少基于与该参考信号比对的多个序列信号、已知序列或其组合来执行。在一些实施方案中,该比对可以包括执行一种或多种模拟信号处理算法。
[0122]
在一些实施方案中,量化多个序列信号的邻近序列依赖性包括对包含未知序列的第二集合的dna分子进行测序,从而生成第二多个序列信号。第二多个序列信号可以与参考信号(例如,类似物参考信号)进行比对。在第二多个序列信号进行比对后,可以确定第二多个dna分子的碱基判定。这样的确定可以至少基于与该参考信号比对的多个序列信号、量化的邻近序列依赖性或其组合。
[0123]
在一些实施方案中,dna分子衍生自核糖核酸(rna)分子。例如,可以通过对rna分子执行逆转录以生成互补dna(cdna)分子或其衍生物来生成dna分子。多个序列信号和/或估算序列可以通过任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。在一些实施方案中,量化邻近序列依赖性包括针对多个位点中的每一
个建立信号幅度与碱基判定和/或序列长度之间的关系。这样的关系可以表示为邻近序列特异性映射(邻近序列图谱)。
[0124]
本公开内容的方法可以包括处理多个序列信号。该方法可包括对核酸样品进行测序以提供多个序列信号。多个序列信号可以与参考信号(例如,类似物参考信号)进行比对。在多个序列信号与参考信号进行比对后,可鉴别包含碱基序列的基因组位点。可以至少基于比对的序列信号来执行该鉴别。可对与参考信号比对的多个序列信号进行处理以鉴别碱基序列的碱基判定和/或估计长度。可使用一种或多种算法通过将信号转换为碱基判定和序列长度(例如,使用邻近序列图谱或其他邻近序列依赖性信息)来鉴别碱基序列的碱基判定和/或估计长度。序列的估计碱基判定和序列长度可使用二次分析数据进行优化。这样的二次分析数据可用于提供或增强邻近序列依赖性信息。多个序列信号可以由任何合适的测序方法如大规模平行阵列测序、流式测序、边合成边测序或染料测序来生成。计算机系统
[0125]
本公开提供了被编程为实现本公开的方法的计算机控制系统。图5示出了计算机系统501,其被编程或以其他方式配置用于例如:生成用于对核酸分子进行条形码化的条形码集;对条形码化核酸分子进行测序以生成包含对应于条形码序列的信号的测序信号;和/或使用对应于条形码序列的信号将测序信号分组成组,其中给定组的测序信号包括对应于条形码序列中(i)对于给定组是相同的并且(ii)与其他组的条形码序列不同的条形码序列的信号;处理给定组内的测序信号以生成聚集信号集,将该聚集信号集组合以生成共有序列。
[0126]
计算机系统501可以调节本公开内容的方法和系统的各个方面,例如,生成用于对核酸分子进行条形码化的条形码集;对条形码化核酸分子进行测序以生成包含对应于条形码序列的信号的测序信号;使用对应于条形码序列的信号将测序信号分组成组,其中给定组的测序信号包括对应于条形码序列中(i)对于给定组是相同的并且(ii)与其他组的条形码序列不同的条形码序列的信号;处理给定组内的测序信号以生成聚集信号集,将该聚集信号集组合以生成共有序列。
[0127]
计算机系统501可以是用户的电子设备或相对于电子设备远程定位的计算机系统。该电子设备可以是移动电子设备。计算机系统501包括中央处理单元(cpu,本文也称为“处理器”和“计算机处理器”)505,其可以是单核或多核处理器,或者是用于并行处理的多个处理器。计算机系统501还包括存储器或存储器位置510(例如,随机存取存储器、只读存储器、闪存)、电子存储单元515(例如,硬盘)、用于与一个或多个其他系统通信的通信接口520(例如,网络适配器)以及外围设备525,诸如高速缓存、其他存储器、数据存储和/或电子显示适配器。存储器510、存储单元515、接口520和外围设备525通过诸如主板等通信总线(实线)与cpu 505通信。存储单元515可以是用于存储数据的数据存储单元(或数据存储库)。计算机系统501借助于通信接口520可操作地耦合到计算机网络(“网络”)530。网络530可以是因特网、互联网和/或外联网,或者与因特网通信的内联网和/或外联网。在一些情况下,网络530是电信和/或数据网络。网络530可以包括一个或多个计算机服务器,其可以实现分布式计算,诸如云计算。在一些情况下,网络530可以借助于计算机系统501实现对等网络,这可以使得耦合到计算机系统501的设备能够起到客户端或服务器的作用。
[0128]
cpu 505可以执行一系列机器可读指令,该机器可读指令可以体现在程序或软件
中。指令可以存储在存储位置如存储器510中。指令可以针对cpu 505,该指令随后可以编程或以其他方式配置cpu 505以实现本公开的方法。由cpu 505执行的操作的实例可以包括提取、解码、执行和回写。
[0129]
cpu 505可以是电路如集成电路的一部分。电路中可以包括系统501的一个或多个其他组件。在一些情况下,该电路是专用集成电路(asic)。
[0130]
存储单元515可以存储文件,诸如驱动程序、库和保存的程序。存储单元515可以存储用户数据,例如用户偏好和用户程序。在一些情况下,计算机系统501可以包括一个或多个附加数据存储单元,所述附加数据存储单元位于计算机系统501外部,诸如位于通过内联网或因特网与计算机系统501通信的远程服务器上。
[0131]
计算机系统501可通过网络530与一个或多个远程计算机系统通信。例如,计算机系统501可以与用户的远程计算机系统通信。远程计算机系统的示例包括个人计算机(例如,便携式pc)、平板或平板型pc(例如,galaxytab)、电话、智能电话(例如,支持android的设备、)或个人数字助理。用户可以经由网络530访问计算机系统501。
[0132]
本文所述的方法可通过机器(例如,计算机处理器)可执行代码的方式来实现,该机器可执行代码存储在计算机系统501的电子存储位置上,例如存储器510或电子存储单元515上。机器可执行代码或机器可读代码可以以软件的形式提供。在使用期间,该代码可由处理器505执行。在一些情况下,可从存储单元515检索代码并将其存储在存储器510上,以供处理器505迅速存取。在一些情况下,可排除电子存储单元515,并且将机器可执行指令存储在存储器510上。
[0133]
该代码可以被预编译并配置用于由具有适于执行该代码的处理器的机器使用,或者可以在运行期间被编译。代码可以用编程语言提供,可以选择编程语言以使代码能够以预编译或即时编译(as-compiled)的方式执行。
[0134]
本文提供的系统和方法的各个方面,诸如计算机系统501,可以在编程中体现。该技术的各个方面可以被认为是“产品”或“制品”,其一般为在一种类型的机器可读介质中携带或体现的机器(或处理器)可执行代码和/或相关数据的形式。机器可执行代码可以存储在电子存储单元如存储器(例如,只读存储器、随机存取存储器、闪存)或硬盘上。“存储”型介质可以包括计算机的任何或全部有形存储器、处理器等,或其相关模块,诸如各种半导体存储器、磁带驱动器、磁盘驱动器等,其可以在任何时间为软件编程提供非暂时性存储。软件的全部或部分有时可以通过因特网或各种其他电信网络进行通信。例如,这样的通信可以使软件从能够一台计算机或处理器加载到另一台计算机或处理器中,例如从管理服务器或主机加载到应用服务器的计算机平台中。因此,可以承载软件元素的另一类型的介质包括光波、电波和电磁波,诸如跨本地设备之间的物理接口、通过有线和光学陆线网络以及各种空中链路而使用的。携载此类波的物理元件,诸如有线或无线链路、光学链路等,也可以被视为承载软件的介质。如本文所用,除非限于非暂时性有形的“存储”介质,否则诸如计算机或机器“可读介质”等术语是指参与向处理器提供指令以供执行的任何介质。
[0135]
因此,机器可读介质如计算机可执行代码可采取多种形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括例如光盘或磁盘,诸如任何计算
机中的任何存储设备等,诸如可用于实现如附图中所示的数据库等。易失性存储介质包括动态存储器,诸如这样的计算机平台的主存储器。有形传输介质包括同轴缆线、铜线和光纤,包括构成计算机系统内的总线的线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频(rf)和红外(ir)数据通信期间产生的那些。因此,计算机可读介质的常见形式包括例如:软盘、柔性盘、硬盘、磁带、任何其他磁性介质、cd-rom、dvd或dvd-rom、任何其他光学介质、穿孔卡片纸带、任何其他具有孔洞图案的物理存储介质、ram、rom、prom和eprom、flash-eprom、任何其他存储器芯片或匣盒、传送数据或指令的载波、传送此类载波的缆线或链路,或者计算机可以从中读取编程代码和/或数据的任何其他介质。这些形式的计算机可读介质中的许多介质可以参与将一个或多个指令的一个或多个序列携载到处理器以供执行。
[0136]
计算机系统501可以包括电子显示器535,或者与电子显示器535通信,电子显示器535包括用于提供例如算法、信号数据、序列数据和数据库的用户选择的用户界面(ui)540。ui的实例包括但不限于图形用户界面(gui)和基于web的用户界面。
[0137]
本公开内容的方法和系统可通过一种或多种算法来实现。算法可以在由中央处理单元505执行时通过软件的方式来实现。算法可以例如生成用于对核酸分子进行条形码化的条形码集;对条形码化核酸分子进行测序以生成包含对应于条形码序列的信号的测序信号;使用对应于条形码序列的信号将测序信号分组成组,其中给定组的测序信号包括对应于条形码序列中(i)对于给定组是相同的并且(ii)与其他组的条形码序列不同的条形码序列的信号;处理给定组内的测序信号以生成聚集信号集,将该聚集信号集组合以生成共有序列。集成测序信号以实现准确的碱基判定
[0138]
如图1所示,可以使用原始测序信号(例如,每个流循环期间的荧光测量)作为对测序数据进行准确分组的基础。特别地,原始信号提供了使用分析方法(诸如信号平均)来减少或消除系统误差的可能性。因此,基于原始信号的分类可能更准确。作为说明,图6-9呈现了示例。数据平均技术可应用于原始测序数据,从而在多个模板分子之间实现更准确的碱基判定。当不同的神经网络模型用于碱基判定时,观察到类似的结果。
[0139]
在一些实施方案中,可在分析的不同阶段对原始信号应用平均技术(其中待平均的原始信号的数量可以变化,例如,10倍、100倍、1000倍、10,000倍或更多倍)。然后可以将平均信号用作用于碱基判定的训练模型的输入(例如,人类基因组训练神经网络模型或大肠杆菌基因组训练神经网络模型)。在一些实施方案中,仍可将原始信号提供给用于碱基判定的训练模型,但可以对碱基判定模型的输出进行平均。例如,训练模型可以输出多个概率(例如,4个概率),每个概率对应于基于来自与特定模板杂交的珠的数据出现在给定位置的特定碱基类型的可能性。然后可以对从与同一模板杂交的多个珠计算的输出概率进行平均。在一些实施方案中,可以以多个水平应用平均技术。例如,可以对与相同模板分子杂交的每十个珠的原始信号进行平均并将平均数据用作用于碱基判定的训练模型的输入,此外,碱基判定模型的输出可以在十个珠子的不同组中进行平均(例如,每十个珠都可以视作一个超级珠)。
[0140]
虽然所描述的分析可以结合模板分子执行,但类似的方法也可以结合条形码序列或信号分组和子分组分析执行(例如,如图1所示)。例如,以下实施例中的每个模板分子(或
其一部分)都可以被认为是一个条形码。应用本文公开内容的方法可以导致基于条形码序列的更准确的分组。附加地,如果将模板分子的一部分视作条形码,则也可以将模板分子序列的其余部分视作靶分子(例如,进行变体分析的分子)。更准确的条形码组与更准确的靶区域碱基判定相结合可以提高变体鉴别的准确性。实施例
[0141]
实施例1:
[0142]
使用本公开内容的方法和系统,若干已知模板的测序数据用于证明通过多个平均技术(例如,平均测序信号从而创建“超珠”,在碱基判定之前平均来自碱基判定算法的输出,通过平均技术的组合等)执行改进的碱基判定的有利效果。可以在不使用分子条形码来区分来自多个模板分子中的各个模板分子的情况下进行这样的分析。性能分析包括,对于多个模板分子中的每一个,将对与多个模板分子相关的超珠执行的碱基判定(例如,使用一个或多个平均技术)的错误率与对基于来自与多个模板分子相关的多个珠的输入执行的碱基判定(例如,不进行平均)的错误率进行比较。
[0143]
在一些实施方案中,为特定实验选择模板分子(例如,从tf1l、tf2l、tf3l、tf4l、tf5l、tf6l等中选择)。接下来,收集模板分子的测序数据;例如,来自多个珠,每个珠带有模板分子。接下来,使用神经网络模型(例如,在人类基因组、大肠杆菌基因组或另一参考基因组上训练),对来自与相同模板分子杂交的每个珠的多个单独模板读取进行碱基判定,从而确定模板分子的序列信息。接下来,跨分析中包含的多个珠确定每个模板的错误率(例如,使用单次运行)。
[0144]
在一些实施方案中,对于给定模板类型,将给定模板类型的多个珠的信号平均在一起以创建“超珠”。例如,可通过对来自约5个珠、约10个珠、约20个珠、约30个珠、约40个珠、约50个珠、约60个珠、约70个珠、约80个珠、约90个珠、约100个珠、约200个珠、约300个珠、约400个珠、约500个珠、约600个珠、约700个珠、约800个珠、约900个珠、约1000个珠、约2000个珠、约3000个珠、约4000个珠、约5000个珠、约6000个珠、约7000个珠、约8000个珠、约9000个珠、约10000个珠等的信号进行平均以生成“超珠”。接下来,使用相同的人类基因组训练神经网络模型对超珠执行碱基判定。接下来,确定超珠的错误率并将其与每个模板的错误率进行比较,从而确认使用超珠的碱基判定的信号平均技术降低了错误率。
[0145]
在一些实施方案中,在确认信号平均技术导致证实所有珠的性能改进后,针对更少数量的珠的给定的模板分子重复实验(例如,对约5个珠、约10个珠、约20个珠、约30个珠、约40个珠、约50个珠、约60个珠、约70个珠、约80个珠、约90个珠、约100个珠、约200个珠、约300个珠、约400个珠、约500个珠、约600个珠、约700个珠、约800个珠、约900个珠、约1000个珠、约2000个珠、约3000个珠、约4000个珠、约5000个珠、约6000个珠、约7000个珠、约8000个珠、约9000个珠、约10000个珠等的组之间的信号进行平均)。
[0146]
选择另一模板分子时,可以使用不同的模板分子重复实验。
[0147]
对6个标准模板分子tf1l、tf2l、tf3l、tf4l、tf5l和tf6l中多个的每一个进行实验。此外,使用两个单独训练的神经网络模型执行碱基判定实验:在人类基因组上训练的第一神经网络模型(例如,人hg或nn模型)并且在大肠杆菌基因组上训练的第二神经网络(例如,大肠杆菌nn模型)。
[0148]
图6示出了tf1l模板的碱基判定分析的示例。此处,对每个流循环的荧光信号进行
量化,在此期间特定类型的核苷酸可被延伸的模板分子接近。使用人类基因组训练的神经网络模型执行碱基判定。上图图示了随机选择的珠的碱基判定结果,每个珠都与tf1l模板杂交,不进行信号平均。指示真实模板序列的真实密钥(true-key)如黑色圆圈所示。为简单起见,描述了单个珠的碱基判定结果,但未指定碱基类型。如图所示,不同珠的碱基判定结果以相当大的波动分散在每个循环中。下图图示了使用信号平均技术的碱基判定结果;例如,基于100个平均信号,每个信号在随机选择的10个珠的多数上测量,每个珠与tf1l模板杂交。“全平均”图描绘了当信号在大量珠(例如,几万个珠)上平均后的神经网络预测。备选地,可基于神经网络模型的输出计算平均。仍备选地,可以使用组合的平均方法。例如,可以平均每组珠的荧光信号(例如,每组包含10至100个珠)。然后将平均信号用作用于碱基判定的预训练神经网络模型的输入。来自神经网络模型的输出(例如,每个代表特定碱基类型存在于模板中特定位置的可能性的概率值)可以在特定位置的最终碱基判定之前进一步平均。
[0149]
上图显示,在不进行平均的情况下,来自随机选择的珠的信号分散在周围,有时会显著偏离真实秘钥碱基类型。相比之下,平均信号始终导致准确的碱基判定,与真实key中的碱基判定一致。
[0150]
图7示出了tf4l模板的碱基判定分析的示例。此处,对每个流循环的荧光信号进行量化,在此期间特定类型的核苷酸可被延伸的模板分子接近。使用人类基因组训练的神经网络模型执行碱基判定,并以类似于图6中的方式呈现数据。观察到类似的结果。图7的上图显示,在不进行平均的情况下,来自随机选择的珠的信号分散在周围,有时会显著偏离真实key碱基类型。相比之下,平均信号始终导致准确的碱基判定,与真实key中的碱基判定一致。
[0151]
图8示出了tf3l模板的碱基判定分析的示例,使用用于碱基判定的大肠杆菌基因组训练神经网络模型。图9示出了tf4l模板的碱基判定分析的示例,使用用于碱基判定的大肠杆菌基因组训练神经网络模型。在图8-图9所示的两个实验中观察到与使用预训练的人类神经网络模型观察到的结果相似的结果。在不进行平均的情况下,来自随机选择的珠的信号分散在周围,有时会显著偏离真实key碱基类型。相比之下,平均信号始终导致准确的碱基判定,与真实key中的碱基判定一致。
[0152]
表1示出了使用不同模板分子(例如,phix-2941l、tf1l、tf3l、tf4l、tf5l和tf6l)和使用不同神经网络模型(例如,人nn模型和大肠杆菌nn模型)的各种珠判定实验获得的珠错误率(ber)汇总。
[0153]
表1:使用人和大肠杆菌nn模型的模板分子中的珠错误率
[0154]
如图6-图9和表1所示,报告了这6个标准模板分子的实验结果,包括使用各种技术
的标准6个模板的珠错误率(ber),包括每个珠的所有单个错误的碱基判定,对10个珠进行信号平均的碱基判定,对100个珠进行信号平均的碱基判定,对1000个珠进行信号平均的碱基判定,对所有珠进行信号平均的碱基判定。特别地,结果表明,对于大多数模板,使用信号平均技术执行碱基判定通常会降低ber(尽管在少数情况下,由于系统误差,ber没有得到改善)。因此,从实验中获得的数据清楚地表明,在某些情况下,使用信号平均技术执行碱基判定可有效降低由于信噪比(snr)增加导致的ber。snr的这样的改进是通过对随机误差产生的“噪声”进行有效的误差抑制来实现的。snr的这种改进例如在模板tf1l、tf3l和tf4l中特别明显。此外,nn模型校正了信号中的一些可变性(例如,跨晶原可变性和对拷贝数的非线性依赖性),从而提高了碱基判定的snr。
[0155]
尽管本文已经示出并描述了本发明的优选实施方案,但对本领域技术人员而言明显的是,这些实施方案仅以示例的方式提供。本发明不意在受说明书中提供的具体示例的限制。虽然已经参考前述说明书描述了本发明,但是对本文的实施方案的描述和说明并非意在以限制性的意义解释。在不偏离本发明的情况下,本领域技术人员现将想到许多变化、改变和替代。此外,应当理解,本发明的所有方面不限于本文阐述的取决于各种条件和变量的特定描述、配置或相对比例。应当理解,本文所述的本发明实施方案的各种替代方案可用于实践本发明。因此,考虑到本发明还应涵盖任何此类替代、修改、变化或等同方案。以下权利要求旨在限定本发明的范围,并由此涵盖这些权利要求范围内的方法和结构及其等同方案。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。