1.本发明涉及化学合成技术领域,具体而言,涉及一种甲硝唑连续化合成方法。
背景技术:
2.甲硝唑(2-甲基-5-硝基-1h-咪唑-1-乙醇)是一种抗生素和抗原虫剂。主要用于治疗或预防厌氧菌引起的系统或局部感染,如腹腔、消化道、女性生殖系、下呼吸道、皮肤及软组织、骨和关节等部位的厌氧菌感染,对败血症、心内膜炎、脑膜感染以及使用抗生素引起的结肠炎也有效。治疗破伤风常与破伤风抗毒素(tat)联用。还可用于口腔厌氧菌感染。
3.甲硝唑的常规生产方法是由2-甲基-5-硝基咪唑与环氧乙烷加成而得。将2-甲基-5-硝基咪唑溶解于甲酸中,在30-40℃逐次加入环氧乙烷,并在加料中间加硫酸。加毕,反应1h。减压回收甲酸,加水溶解后冷却至10℃,过滤。滤液用氢氧化钠溶液调至ph=10,放置冷却,过滤,水洗至近中改建。用水重结晶。活性炭脱色,即得甲硝唑。
4.然而由于环氧乙烷在低温下为无色透明液体,在常温下为无色刺激性气味气体,不易储存。在用环氧乙烷做羟乙基化反应时,环氧乙烷必须液化,采用分布式进料,过程中容易汽化,造成进料不易及进料量不准确。
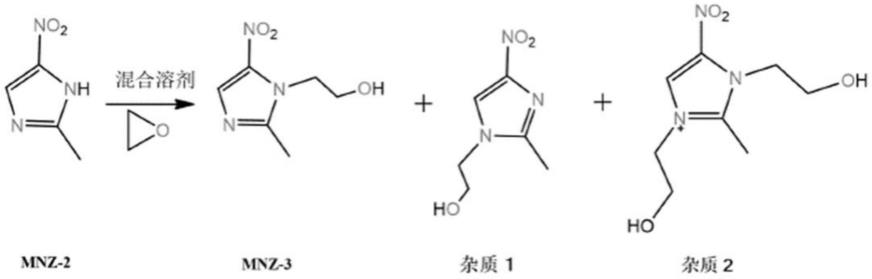
5.并且在由2-甲基-5-硝基咪唑与环氧乙烷加成反应时还容易产生杂质:
[0006][0007]
根据实验现象及文献报道可知,杂质1在碱性环境中非常容易产生(如在四丁基溴化铵催化条件下);杂质2与温度过高、环氧乙烷当量过大、反应时间过长均易产生,2-甲基-5-硝基咪唑的羟乙基化反应属于放热反应,在通入环氧乙烷速率过快或冷却失效条件下,体系内温度过剧烈升高,直至原料完全消耗毕,导致易产生杂质2。
[0008]
因此,采用2-甲基-5-硝基咪唑与环氧乙烷反应制备甲硝唑存在进料不易及进料量不准确且杂质含量较高的情况。
[0009]
鉴于此,特提出本发明。
技术实现要素:
[0010]
本发明的目的在于提供一种甲硝唑连续化合成方法。
[0011]
本发明是这样实现的:
[0012]
第一方面,本发明提供一种甲硝唑连续化合成方法,其包括:将含有2-甲基-5-硝
基咪唑和2-氯乙醇的酸性溶解液通入羟乙基化微通道反应器内经加热发生羟乙基化反应,反应完全后对从所述羟乙基化微通道反应器内流出的反应液进行降温,调节所述反应液的ph至3-4,固液分离取滤液并调节所述滤液的ph值至9-11,析晶得到甲硝唑粗品,重结晶得到甲硝唑。
[0013]
本发明具有以下有益效果:
[0014]
本技术提供的甲硝唑连续化合成方法采用2-甲基-5-硝基咪唑和2-氯乙醇在羟乙基化微通道反应器内进行羟乙基化反应以制备甲硝唑,2-甲基-5硝基咪唑和2-氯乙醇在酸性条件下呈溶解澄清状态,利于进料及准确计量。同时,含有2-甲基-5硝基咪唑和2-氯乙醇的酸性溶解液可以一并通入羟乙基化微通道反应器中,无需将2-甲基-5硝基咪唑和2-氯乙醇单独通入或者多股通入,进料更方便,此外,羟乙基化微通道反应器的反应比表面积大,换热效率高,可以增压提高反应温度,反应完以后,将剩余2-氯乙醇减压蒸出,用于下批投料,避免体系中有大量的酸造成后期调ph时产生大量的盐及废水。
附图说明
[0015]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
[0016]
图1为本技术提供的甲硝唑连续化合成方法中合成2-甲基咪唑的设备流程图;
[0017]
图2为本技术提供的甲硝唑连续化合成方法中硝化反应合成2-甲基-5-硝基咪唑的设备流程图;
[0018]
图3为本技术提供的甲硝唑连续化合成方法中硝化反应合成2-甲基-5-硝基咪唑的硝化微反应器的结构示意图;
[0019]
图4为本技术提供的甲硝唑连续化合成方法中羟乙基化反应的设备流程图;
[0020]
图5为本技术提供的甲硝唑连续化合成方法中羟乙基化反应的羟乙基化微反应器的结构示意图。
[0021]
图标:101-氨水桶;102-醛水桶;103-称重模块;104-进料泵;105-混合器;106-加热恒温槽;107-第一管式反应器;108-冷却管;109-低温恒温槽;110-第一背压阀;111-气液分离器;112-硫酸吸收桶;
[0022]
201-第一柱塞泵;202-第二柱塞泵;203-硝化微反应器;204-进料分布器;205-第二管式反应器;206-硝化接收釜;207-硝化中和釜;208-第二背压阀;209-尾气吸收罐;2031-第二反应预热通道;2032-第二反应通道;
[0023]
301-溶解釜;302-柱塞泵;303-羟乙基化微通道反应器;304-浓缩釜;305-羟乙基化接收釜;306-第一羟乙基化中和釜;307-第一压滤器;308-第二羟乙基化中和釜;309-第二压滤器;3031-第一反应预热通道;3032-第一反应通道。
具体实施方式
[0024]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建
议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0025]
本发明提供一种甲硝唑连续化合成方法,其包括如下反应:
[0026]
s1、合成2-甲基咪唑。
[0027]
合成设备流程图请参阅图1:
[0028]
将乙二醛和乙醛的醛混合液以及氨水混合后,预冷到10-20℃,进入sk型管式反应器于1.8-2.0mpa下进行反应,反应液温度为55-60℃;反应时间为18-22min;反应结束后降温至10-15℃,接着进行浓缩,浓缩的温度为58-62℃,浓缩时间为1-3h,接着结晶;过滤洗涤得到2-甲基咪唑。
[0029]
具体来说,本技术中,乙醛和乙二醛在20℃以下预先混匀后置于醛水桶102中,与氨水桶101内的氨水溶液经称重模块103精准计量后由进料泵104分别送入夹套混合器105中,预冷到10-20℃,再进入扇齿混合器,再次混合后,进入φ8 50m内置sk型的第一管式反应器107反应,管式反应器油温采用加热恒温槽106进行控制在50-60℃。反应液温度控制在55-60℃,反应后的反应液通入低温恒温槽109的冷却管108中进行冷却,冷却后通入气液分离器111中进行气液分离,溢出的气体可以经第一背压阀110排至硫酸吸收桶112中进行吸收,随后对气液分离器111内的反应液进行浓缩结晶和洗涤过滤。
[0030]
当乙醛的当量略微过量,有助于反应的进行。由于氨水中游离氨受热时容易挥发,氨水需大大过量。乙二醛当量为1:1时,2-甲基咪唑收率为75%;继续增加乙醛的当量,当达到1.2eq时,2-甲基咪唑收率高达86%以上。当继续增加乙醛的当量时,收率增加不明显。
[0031]
本技术研究发现,在较低温度时,提高反应温度可以缩短反应停留时间,但是反应温度对杂质的形成有较大的影响,为了提高收率,提高反应温度和改变反应停留时间,当温度达到60℃、停留时间为20min时,理论收率高达93%,当温度超过60℃时,温度越高,焦油状杂质越多,收率越低;当停留时间为10min、温度为70-80℃时,理论收率最高为73%,当停留时间为15min、温度70-80℃时,理论收率最高为79%。因此,本技术中限定在sk型管式反应器中反应液的反应温度为55-60℃;反应时间为18-22min。
[0032]
由于反应体系浓缩前并未淬灭,浓缩时反应仍在进行中,高温度长时间浓缩会导致反应杂质增加,反应液颜色越深,收率变低。经实验发现:浓缩的温度为58-62℃,浓缩时间为1-3h内收率和纯度较佳,优选地,浓缩温度60℃、浓缩时间为3小时内收率和纯度最佳,同时料液颜色最浅。
[0033]
结晶包括向浓缩液中加入氢氧化钠,冷却至35-45℃,逐批加入氯化钠,至有固体析出后,缓慢降温至0-2℃,搅拌25-35min过滤,滤饼用0-2℃的nacl或naoh溶液洗涤,滤饼烘干即得2-甲基咪,收率》85%,hplc纯度》99%,颜色浅黄色。
[0034]
s2、硝化反应合成2-甲基-5-硝基咪唑
[0035]
合成设备流程图请参阅图2和图3:
[0036]
将混合有2-甲基咪唑和硫酸的咪唑硫酸溶液和硝酸分别利用第一柱塞泵201和第二柱塞泵202通入硝化微反应器203中进行硝化反应10-18min,从硝化微反应器203中流出的反应液进入第二管式反应器205中继续反应30-50min,反应完成后将反应体系通入硝化接收釜206进行冷却至室温,冷却后的反应液通入硝化中和釜207中用冰水淬灭,加入碱液调节ph至3.5-4.5,常温搅拌15-25min,接着过滤取滤饼进行漂洗后干燥,得到2-甲基-5-硝
基咪唑,硝化接收釜206中溢出的气体经第二背压阀208通入尾气吸收罐209中进行吸收。
[0037]
本技术中,硝化微反应器203包括一片第二反应预热通道2031和多片依次连通的第二反应通道2032,第二反应预热通道2031与第二反应通道2032连通,第二反应预热通道2031和第二反应通道2032均设置有进料口;将咪唑硫酸溶液送入第二反应预热通道2031内,硝酸分为多股经进料分布器204分别进入多片第二反应通道2032,第二反应预热通道2031内的咪唑硫酸溶液温度达到110℃后进入第一片第二反应通道2032和第一股硝酸混合反应,反应混合物流出第一片第二反应通道2032后,进入第二片第二反应通道2032和第二股硝酸混合反应,以此类推,直至反应混合液从最后一片第二反应通道2032中流出。
[0038]
优选地,硝化微反应器203采用油浴加热,其外温为110-120℃,其内温为120-140℃;由于硝化反应本身为放热反应,因此硝化微反应器203的内温会高于外温。反应通道为5-15片,硝酸对应分为5-15股进料。
[0039]
现有技术中硝化反应通常在釜式反应器中进行,其主反应式为:
[0040][0041]
在主反应的过程中,还存在杂质及其相互转化:
[0042][0043]
根据实验现象及文献报道可知,杂质ip-2a非常容易产生。当硝酸在较低温度下硝化(小于80℃),或者使用较稀浓度的硝酸、稀硝酸、硝酸-醋酐-醋酸体系硝化,或者滴加速度过快,反应体系中ip-2a为主要产物。ip-2a在高温条件下可以转化为目标产物mnz-2。或者在硝化体系中进一步转化为杂质ip-2b。杂质ip-2b在高温条件下可以部分转化为产品mnz-2和杂质ip-2c。当反应体系中硝酸过量时,mnz-2可以进一步硝化为二硝化产物ip-2b和ip-2c。并且所有硝化产物都可以在硝硫混酸硝化体系中高温分解为hplc-uv条件无法检测的小分子。根据此种原理,在硝化反应结束后,高温条件下维持适当时间的搅拌,可以提高反应体系的色谱纯度,但同时mnz-2会随之降低。高温条件下各个杂质的稳定性顺序为ip-2c》ip-2b》ip-2a。硝化反应中产生的杂质也可以通过重结晶的方法来去除,在酸性ph条件下,2a、2b、2c的水溶性都远大于mnz-2。因此ph=4条件下结晶可以去除大部分反应杂质。
[0044]
由于2-甲基咪唑的硝化反应属于强放热反应,在滴加速度过快或冷却失效条件下,体系内温会剧烈升高,直至超过2-甲基-5-硝基咪唑的分解温度(dsc测得其起始分解温度为260℃),进而引起爆炸。在釜式反应中存在的困难在于,如果为了保证安全而过度降温,如反应温度低于120℃,反应体系中副反应特别是ip-2a和ip-2b将是主要产物。因此工业生产过程中,釜式反应器中内温达到130℃以上才可以滴加硝酸,但此时反应热很难有效释放,具有很大失控飞温的风险,并且高温下硝酸分解为氮氧化物,车间操作工况很差。为了抑制温度过快上升,大部分生成工艺中在反应体系中加入了大量的硫酸盐,提高反应体系的比热容;并且已见报道的部分工艺中,尿素也被加入反应体系中来消除氮氧化物,改善反应工况,抑制可能存在的亚硝化反应。但此种方法目前的环保压力较大,工艺末端产生的大量固体废弃物无法处理,这已经严重限制了该方法的应用。
[0045]
本技术创新性的采用硝化微反应器203和第二管式反应器205相结合的反应来进行硝化反应,相较于传统的釜式反应器而言,本技术具有更高的比换热面积,本技术中在强放热和浓酸阶段采用硝化微反应器203,2-甲基咪唑预先溶于浓硫酸中,由第一柱塞泵201送入第二反应预热通道2031,硝酸须分作多股分别进入第二反应通道2032不同位置的进料点,硝酸进料点越多,杂质越少,体系温度越平稳。其中硝酸作1股或者2股进料,反应主要产生副产物ip-2a和ip-2b;当硝酸分为5-15股(优选为10股)进入混合器,反应体系后处理前的液相色谱纯度可达84%以上。在稀酸加热阶段采用第二管式反应器205继续进行反应,可以延长反应时间,提升收率。
[0046]
优选地,在反应混合液从第二管式反应器205中排出后,冷却至室温后加入冰水淬灭,接着再加入饱和氢氧化钠溶液调节体系ph至4-4.5时,停止加碱。值得注意的是,此时继续加入液碱仍然有固体析出,但是大部分是杂质和无机盐。本技术中的氢氧化钠不能用氢氧化钾替代,因为钾盐溶解度较差,大部分会析出和硝基咪唑混合在一起,而钠盐析出较少。
[0047]
s3、羟乙基化反应。
[0048]
其合成反应式如下:
[0049][0050]
其合成设备流程图请参阅图4和图5:
[0051]
将含有2-甲基-5-硝基咪唑和2-氯乙醇的酸性溶解液经加热发生羟乙基化反应,反应完全后,将反应体系中未反应的2-氯乙醇蒸出并回收利用,接着对反应体系进行降温,调节反应体系的ph至3-4,反应体系降温至0-5℃并搅拌1-2h,过滤,滤饼为未反应的2-甲基-5硝基咪唑,取滤液并调节滤液的ph值至9-11,搅拌析晶后,过滤,得到甲硝唑粗品,重结晶得到甲硝唑。
[0052]
其中,酸性溶解液的制备方法包括:先将2-甲基-5-硝基咪唑与2-氯乙醇分别通入溶解釜301中进行混合,搅拌形成混悬液,接着向溶解釜301内的混悬液中加入干燥氯化氢气体、氯化氢溶液或硫酸溶液;优选地,向混悬液中加入干燥氯化氢气体,经研究发现,向混悬液中加入干燥氯化氢气体反应更加迅速,有利于节约反应时间,本技术中通过限定酸性溶解液的ph为2-3来控制加入干燥氯化氢气体、氯化氢溶液或硫酸溶液的量,或者通过混悬液是否完全溶解来判断加入干燥氯化氢气体、氯化氢溶液或硫酸溶液的量。
[0053]
本技术中,2-甲基-5-硝基咪唑与2-氯乙醇的质量比为1-2:5-8,可以看出,2-氯乙醇采用过量加入的方式,保证2-甲基-5-硝基咪唑尽可能的反应,未反应的2-甲基-5-硝基咪唑与2-氯乙醇均可以回收再利用。
[0054]
值得注意的是,本技术中的酸性溶解液利用柱塞泵302精准进料并通入羟乙基化微通道反应器303内进行羟乙基化反应;羟乙基化微通道反应器303包括一片第一反应预热通道3031和多片依次连通的第一反应通道3032,第一反应预热通道3031与第一反应通道3032连通;将酸性溶解液送入第一反应预热通道3031内,温度达到110℃后进入第一片第一反应通道3032继续进行反应,反应混合物流出第一片第一反应通道3032后,进入第二片第一反应通道3032继续进行反应,以此类推,直至反应混合液从最后一片第一反应通道3032中流出。羟乙基化反应的反应时间为1-2h,反应温度为130-150℃,羟乙基化反应于2-3mpa的压力条件下进行。
[0055]
常规使用2-氯乙醇做羟乙基化反应时存在反应时间长,转化率偏低,本技术创新性的提出了在羟乙基化微通道反应器303内进行羟乙基化反应,可解决此问题,因为微通道的反应比表面积大,换热效率高,可以增压提高反应温度,2-甲基-5硝基咪唑,2-氯乙醇在酸性条件下呈溶解澄清状态,利于进料及准确计量。
[0056]
反应完以后,反应液流出至浓缩釜304中以便于将剩余2-氯乙醇减压蒸出,蒸出的2-氯乙醇被羟乙基化接收釜305进行接收,用于下批投料,同时还可以避免体系中有大量的酸造成后期调ph时产生大量的盐及废水。
[0057]
接着,将反应体系通入第一羟乙基化中和釜306中进行冷却,其中第一羟乙基化中和釜306中装有冰水,并使用高低温一体机进行夹套冷却。当反应液全部用冰水淬灭后,再加入液碱(40wt%),调节ph=3-4,此时有大量固体产生,中和结束后反应体系降温至0-5℃并搅拌1-2h,第一羟乙基化中和釜306中所有操作都配有强搅拌,第一羟乙基化中和釜306中产生的固体进入第一压滤器307中压滤,滤饼用常温水漂洗两次。吹干后进入烘箱80
°
减压烘干过夜,即为未反应的2-甲基-5硝基咪唑。
[0058]
冷却后的反应液进入第二羟乙基化中和釜308中,其中第二羟乙基化中和釜308中装有冰水,并使用高低温一体机进行夹套冷却。当反应液全部用冰水淬灭后,再加入液碱(40wt%),调节ph=10,此时有大量固体产生,中和结束后常温保持搅拌20min。第二羟乙基化中和釜308中所有操作都配有强搅拌,第二羟乙基化中和釜308中产生的固体进入第二压滤器309中压滤,滤饼用常温水漂洗两次。吹干后进入烘箱80
°
减压烘干过夜。
[0059]
对甲硝唑粗品进行重结晶包括:向甲硝唑粗品中加入水和活性炭进行重结晶;甲硝唑粗品、水和活性炭的质量比为1:4-6:0.4-0.6。
[0060]
需要进一步说明的是,本技术中的步骤s1和步骤s2可以省略,也即是,本技术可以直接采用市售的2-甲基-5-硝基咪唑与2-氯乙醇进行反应,无需对2-甲基-5-硝基咪唑进行
预先合成。
[0061]
本技术中提及的羟乙基化微通道反应器303和硝化微反应器203的结构基本与现有技术中的微通道反应器结构相同,本技术不做具体的限制。本技术中仅仅是利用微通道反应器进行特定的反应,通过羟乙基化微通道反应器303和硝化微反应器203具有的反应比表面积大,换热效率高,可以增压提高反应温度等特点来提升反应速度,提高收率和纯度。
[0062]
以下结合实施例对本发明的特征和性能作进一步的详细描述。
[0063]
实施例1:合成2-甲基咪唑。
[0064]
将10kg 40%乙二醛和9.1kg 40%乙醛在20℃以下预先混合后,与氨水溶液经进料泵分别送入夹套混合器中,预冷到15℃,再进入扇齿混合器,再次混合后,进入φ8 50m内置sk型的第一管式反应器107于2.0mpa下进行反应,第一管式反应器107的油温控制在55℃,反应液温度控制在55℃,反应时间为18-22min。反应液从第一管式反应器107流出时,经温度传感(随时监控反应温度)后,进入冷却管108,降温至15℃,反应液经减压浓缩,浓缩的温度为60℃,浓缩时间为2h,降温、析晶、压滤、重结晶、烘干得4.8kg浅黄色固体,纯度为98%,收率为28.4%。
[0065]
实施例2:硝化反应合成2-甲基-5-硝基咪唑
[0066]
将混合有2-甲基咪唑和硫酸的咪唑硫酸溶液以20g/min流速送入第二反应预热通道2031内,硝酸分为10股分别以15g/min流速进入10片第二反应通道2032(内温为120-140℃),第二反应预热通道2031内的咪唑硫酸溶液温度达到110℃后进入第一片第二反应通道2032和第一股硝酸混合反应,反应混合物流出第一片第二反应通道2032后,进入第二片第二反应通道2032和第二股硝酸混合反应,以此类推,直至反应混合液从最后一片第二反应通道2032中流出,反应混合液在硝化微反应器203中的停留时间为15min,从硝化微反应器203中流出的反应液进入第二管式反应器205中继续反应40min,反应完成后将反应体系进行冷却至室温,冷却后的反应液用冰水淬灭,加入饱和氢氧化钠溶液调节ph至4,常温搅拌20min,接着过滤取滤饼进行漂洗后干燥,得到2-甲基-5-硝基咪唑,纯度大于98%,收率52%。
[0067]
实施例3:羟乙基化反应
[0068]
将20g 2-甲基-5硝基咪唑,50g 2-氯乙醇投入溶解釜301中,搅拌使成为悬浮液,通入干燥氯化氢气体使固体物全溶形成酸性溶解液,将酸性溶解液送入羟乙基化微通道反应器303的第一反应预热通道3031内,温度达到110℃后进入第一片第一反应通道3032继续进行反应,反应混合物流出第一片第一反应通道3032后,进入第二片第一反应通道3032继续进行反应,以此类推,直至反应混合液从最后一片第一反应通道3032中流出,反应液在羟乙基化微通道反应器303中的停留时间为2h;将未反应的2-氯乙醇蒸出,然后降温,用氢氧化钠溶液调节ph=3,降温至0℃搅拌1h,抽滤,滤饼为未反应的2-甲基-5硝基咪唑。滤液加入氢氧化钠溶液调节ph=10.0左右,搅拌析晶2h,抽滤,得甲硝唑粗品10.0g,纯度为98%,收率37%。甲硝唑粗品加入纯化水和活性炭重结晶,得甲硝唑8.5g,纯度>99%,收率85%。
[0069]
对比例1
[0070]
本对比例提供了一种采用釜式反应器制备甲硝唑的方法,其包括如下步骤:
[0071]
将2-甲基-5-硝基咪唑在冰水浴冷却条件下溶于甲酸(重量为2-甲基-5-硝基咪唑3.33倍)和98%硫酸(重量为2-甲基-5-硝基咪唑0.5倍)体系中。保持内温低于35℃,控制内
温60℃,滴加环氧乙烷—滴加时间约2小时。滴加完成后,此时hplc中控纯度在50%以上。将此反应液浓缩(回收溶剂),再加入1倍当量冰水,保持内温再25℃,加入40%氢氧化钠水溶液,直至ph为4,有固体析出。继续搅拌40min后,过滤,滤饼容去离子水漂洗2次,烘干后得到类白色固体,即未反应完全的原料:2-甲基-5-硝基咪唑,其hplc》90%。合并滤液和洗涤液,再加入,加入40%氢氧化钠水溶液,直至ph为10,有固体析出,继续搅拌40min后,过滤,滤饼用去离子水漂洗2次,烘干后得白色或类白色固体,即甲硝唑,其hplc纯度大于99%,收率30%。
[0072]
对比例2
[0073]
本对比例提供了一种采用微反应器制备甲硝唑的方法,其包括如下步骤:
[0074]
1.10l搪玻璃釜中投入1.5kg浓硫酸,3.0kg的2-甲基-4(5)-硝基咪唑分批加入10kg甲酸中,溶解过程大量放热,需用冰水冷却,控制内温不超过40℃。
[0075]
2.将上述硝基咪唑硫酸甲酸溶液用柱塞泵以35g/min流速送入反应预热段(第一片微反应器),当温度达到55℃后,进入反应段(第二片微反应器)和第一股环氧乙烷ea(环氧乙烷:ea=1:1)混合反应。反应混合液流出第二片微反应器后,和第二股环氧乙烷ea混合,进入第三片微反应器反应,以此类推。所有11块微反应器模块集成在一起,置于60℃油浴中。
[0076]
3.环氧乙烷ea用柱塞泵送入分布器后,分十股分别进入第二至第十一片微反应器中,和硝基咪唑硫酸甲酸溶液混合反应。
[0077]
4.当反应混合液从第十一片微反应器中流出后,进入50米内径为8mm的管式反应器中继续反应。管式反应器的油浴温度控制在65℃。
[0078]
5.反应液从管式反应器中流出后,进入接收浓缩釜中进行减压浓缩回收甲酸,体系加热至65℃。其中挥发出的甲酸和剩余的环氧乙烷进行酸吸收处理。
[0079]
6.浓缩完毕,冷却后的反应液进入淬灭中和釜中,其中釜中装有冰水3.0kg,并使用高低温一体机进行夹套冷却。当反应液全部用冰水淬灭后,再加入1.0kg液碱(40%),调节ph=4,此时有大量固体产生,中和结束后常温保持搅拌20min。中和釜中所有操作都配有强搅拌。
[0080]
7.中和釜中产生的固体进入压滤器中压滤,滤饼用0.6kg常温水漂洗两次。吹干后进入烘箱80℃减压烘干过夜,得白色或类白色固体,hplc》90%,回收率为30%,即2-甲基-5-硝基咪唑。
[0081]
8.滤液及漂洗液进入下一步中和釜中,继续冷却后的反应液进入淬灭中和釜中,其中釜中装有冰水3.0kg,并使用高低温一体机进行夹套冷却。当反应液全部用冰水淬灭后,再加入1.0kg液碱(40%),调节ph=10,此时有大量固体产生,中和结束后常温保持搅拌20min。中和釜中所有操作都配有强搅拌。
[0082]
9.中和釜中产生的固体进入压滤器中压滤,滤饼用0.6kg常温水漂洗两次。吹干后进入烘箱80℃减压烘干过夜,得白色或类白色固体,即甲硝唑,hplc>99%,收率45%。
[0083]
对比例3
[0084]
本对比例与实施例3基本相同,区别仅在于,将实施例3于反应釜中进行反应,不通入羟乙基化微通道反应器中。制备获得的甲硝唑hplc>99%,收率31%。
[0085]
对比例4
[0086]
本对比例与实施例3基本相同,区别仅在于,将实施例3的2-氯乙醇替换为环氧乙烷。制备获得的甲硝唑hplc>99%,收率44%。
[0087]
对比例5
[0088]
本对比例基本与对比例2相同,区别仅在于,本对比例中,环氧乙烷ea不经分布器分10股进入微通道反应器中,而是直接在进入微通道反应器之前即与步骤1中的硝基咪唑硫酸甲酸溶液混合后,再一并通入微通道反应器中。最终得到的2-甲基-5-硝基咪唑,hplc>90%,收率30%。
[0089]
对比例6
[0090]
本对比例与实施例2基本相同,区别仅在于:省略了实施例2中“从硝化微反应器中流出的反应液进入管式反应器中继续反应30-50min”,也即是,本对比例中,硝化反应仅在硝化微反应器中进行。最终得到的2-甲基-5-硝基咪唑,hplc>99%,收率35%。
[0091]
对比例7
[0092]
本对比例与实施例2基本相同,区别仅在于:将实施例2中“加入饱和氢氧化钠溶液调节ph至4”替换为“加入氨水调节ph至4”。最终得到的2-甲基-5-硝基咪唑,hplc>99%,收率52%。
[0093]
综上所述,本技术提供的甲硝唑连续化合成方法采用2-甲基-5-硝基咪唑和2-氯乙醇在羟乙基化微通道反应器内进行羟乙基化反应以制备甲硝唑,2-甲基-5硝基咪唑和2-氯乙醇在酸性条件下呈溶解澄清状态,利于进料及准确计量。同时,含有2-甲基-5硝基咪唑和2-氯乙醇的酸性溶解液可以一并通入羟乙基化微通道反应器中,无需将2-甲基-5硝基咪唑和2-氯乙醇单独通入或者多股通入,进料更方便,此外,羟乙基化微通道反应器的反应比表面积大,换热效率高,可以增压提高反应温度,反应完以后,将剩余2-氯乙醇减压蒸出,用于下批投料,避免体系中有大量的酸造成后期调ph时产生大量的盐及废水。
[0094]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。