一种mxene基热催化合成氨催化剂及其制备和应用
技术领域
1.本发明涉及催化剂材料制备技术领域,具体地,涉及一种mxene基热催化合成氨催化剂及其制备方法和应用。
背景技术:
2.氨(nh3)是粮食安全的保障,也是高体积能量密度的储氢载体。目前,传统工业合成氨是以化石资源为燃料,采用haber-bosch工艺,h2主要通过水煤气变换反应或者甲烷重整反应产生并采用fe基催化剂,在高温(490~510℃)和高压(15.0~32.0mpa)条件下合成氨每年消耗全球能耗的1-2wt%,且排放大量co2。在节能减排和2060年中国实现“碳中和”的重大长期战略目标下,发展可再生能源驱动的“绿氨”合成显得尤为重要和迫切。我国是可再生能源最大的国家,但我国年“三弃”电量高达约1020亿千瓦时。若以nh3为储氢载体,利用可再生能源电力电解水制氢作为“氢源”,空气分离出氮气作为“氮源”,发展“可再生能源电解水制氢耦合合成氨”技术,可同步实现可再生能源清洁高效利用﹑“绿氨”合成和安全储运氢气。
3.目前,压力型电解水制氢系统要求输出压力≤5.0mpa,一般输出压力为1.6-3.2mpa,电解得到的h2经深度脱水脱氧后的温度约为300-400℃。因此,要实现可再生能源电力和合成氨技术互补融合,亟需发展与可再生能源电力电解制氢体系相匹配的相对温和条件下合成氨技术(反应条件:300~400℃、1.6~3.2mpa),现有面向化石能源的工业合成氨催化剂在此相对温和条件下难以满足要求,设计开发新型高效的合成氨催化剂就成为贯通“可再生能源-氨-氢”循环路线的关键。
4.虽然ru基催化剂相对于fe基催化剂基础上显著降低了合成氨能耗,但ru是贵金属,价格昂贵且地球上储量有限,导致其大规模工业应用具有局限性。因此,要真正实现合成氨过程的进一步大幅度节能降耗,开发低温和低压下高效合成氨催化剂是关键。2016年2月,美国能源部在针对合成氨未来发展战略研讨会上提出,制备出高活性、长寿命的催化剂并实现在温和条件下(200~400℃、0.1~5.0mpa)合成氨是未来的发展方向,如何设计开发出新型非贵金属基高性能催化剂、并实现在温和条件下合成氨成为最具挑战性的研究之一。
技术实现要素:
5.本发明提供一种催化剂,所述催化剂以过渡金属和mo2ct
x
为活性中心,所述过渡金属负载在所述mo2ct
x
上;
6.x代表官能团层数;
7.所述过渡金属选自铁(fe)、钴(co)、镍(ni)、铼(re)中的一种或多种,优选为铼(re)。
8.根据本发明的实施方案,所述过渡金属的负载量为mo2ct
x
的5-15wt%,所述负载量
指过渡金属与所述mo2ct
x
的质量百分比。例如,所述负载量可以为5wt%、6wt%、7wt%、8wt%、9wt%、10wt%、11wt%、12wt%、13wt%、14wt%、15wt%,或者两两组合范围内的任意一个点值。
9.根据本发明的实施方案,所述mo2ct
x
富含表面基团,所述表面基团包括o、oh和f基团中的一种或多种。
10.根据本发明的实施方案,所述mo2ct
x
富含缺陷位,所述缺陷位为金属空位,用于锚定所述过渡金属。例如,所述过渡金属可以以过渡金属单原子、过渡金属纳米颗粒中的一种或多种形式锚定在所述金属空位中。示例性地,所述过渡金属以过渡金属纳米颗粒形式锚定在所述金属空位中。
11.根据本发明的实施方案,所述催化剂的n2反应级数为0.18-0.26,优选为0.18-0.25。
12.根据本发明的实施方案,所述催化剂选自下述催化剂中的任意一种:
13.所述催化剂为re/mo2ct
x
催化剂,以re和mo2ct
x
为活性中心,re至少负载在所述mo2ct
x
的金属空位中;
14.或者,所述催化剂为fe/mo2ct
x
催化剂,以fe和mo2ct
x
为活性中心,fe至少负载在所述mo2ct
x
的金属空位中;
15.或者,所述催化剂为co/mo2ct
x
催化剂,以co和mo2ct
x
为活性中心,co至少负载在所述mo2ct
x
的金属空位中;
16.或者,所述催化剂为ni/mo2ct
x
催化剂,以ni和mo2ct
x
为活性中心,ni至少负载在所述mo2ct
x
的金属空位中。
17.本发明还提供上述催化剂的制备方法,所述制备方法包括以下步骤:
18.(1)使用氢氟酸刻蚀mo2ga2c前驱体材料,获得mo2ct
x
;
19.(2)采用初湿浸渍法,将过渡金属负载到步骤(1)得到的mo2ct
x
上;
20.(3)将步骤(2)中负载过渡金属的mo2ct
x
经加热焙烧还原后,得到所述催化剂。
21.根据本发明的实施方案,步骤(1)中,刻蚀的温度为50-70℃,优选为55℃;刻蚀的时间为3-7天,优选为5天。
22.根据本发明的实施方案,步骤(1)中,采用的氢氟酸可以为氢氟酸溶液;氢氟酸溶液和mo2ga2c前驱体材料的体积质量比为(25-30)ml:1g,示例性为30ml:1g。氢氟酸溶液的浓度为≥40.0wt%。若氢氟酸溶液的浓度和使用量过高,容易导致刻蚀过度,产率较低;若氢氟酸溶液的浓度和使用量过低,导致刻蚀时间太长,或刻蚀不完全。
23.根据本发明的实施方案,步骤(1)中,所述mo2ct
x
具有如上文所示的含义。
24.根据本发明的实施方案,步骤(1)中还包括后处理步骤(1a),刻蚀的反应产物经洗涤、分离、至滤液洗至中性,对刻蚀的反应产物进行干燥;干燥得到的粉末经碱性溶液处理,洗涤,干燥后得到mo2ct
x
。
25.示例性地,干燥得到的粉末与碱性溶液的质量体积比为0.8g:1l。
26.本发明中,氢氟酸处理后,得到mo2ct
x
的表面存在较多f基团,水洗较难洗净。采用碱性溶液清洗能够除去表面f基团,减少f含量,且f、s、cl等元素的存在,对于氨合成反应有抑制作用,易造成催化剂中毒。优选地,所述刻蚀的反应产物的洗涤可以使用去离子水,所述碱性溶液处理后的洗涤可以使用碱性溶液和/或去离子水。例如,洗涤2次。
0.26;优选为0.18-0.25。
44.根据本发明的实施方案,所述合成氨的温和条件包括:温度为300~400℃,压力1mpa。
45.本发明还提供一种合成氨的催化剂,至少含有上述催化剂。
46.本发明还提供一种合成氨的方法,所述合成方法采用上述催化剂。
47.发明人发现,mo2ct
x
作为一种新型二维结构材料,其表面具有较多的负电子基团,给电子能力较强,对n2和h2的吸附能力适中,既可促进n2和h2的活化,又可使氨合成反应中间物种n2h
x
的脱附较为容易,因此,将mo2ct
x
与过渡金属结合形成的复合材料是潜在的氨合成催化剂。本发明提供的mxene基热催化合成氨催化剂及其制备方法和应用,所述催化剂的n2反应级数仅为0.18-0.26,在温和条件下表现出了优异的氨合成性能和长周期催化稳定性。
48.本发明的有益效果:
49.1.本发明通过氢氟酸刻蚀和初湿浸渍的方法,合成了mo2ct
x
负载不同过渡金属温和合成氨催化剂,为过渡金属温和条件下热催化合成氨催化剂的研究和使用提供了解决方案。
50.2.本发明制备的催化剂,通过调控过渡金属的种类,制备温和条件下具有较高活性的合成氨催化剂。
51.3.本发明制备的mxene基热催化合成氨催化剂,较传统的ru和fe基催化剂具有优异的合成氨反应速率,且热稳定性好。本发明提供的催化剂制备方法较为简便,催化剂易于成型,有利于工业应用且大大降低成本。
附图说明
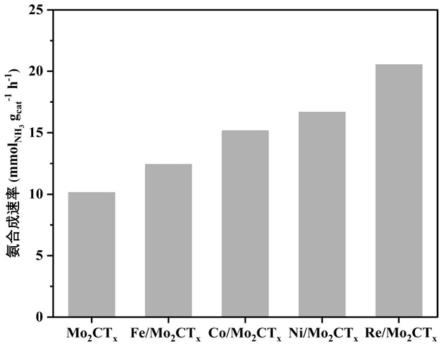
52.图1为对比例1及实施例1-4所得催化剂的合成氨性能图;
53.图2为对比例1及实施例1所得催化剂的n2反应级数图;
54.图3为对比例1及实施例1所得催化剂的活化能图;
55.图4为对比例1及实施例1所得催化剂在400℃,1mpa下的热稳定性图;
56.图5为对比例1所得催化剂在400℃,1mpa下的甲烷化实验测试结果图。
57.图6为对比例1及实施例1所得催化剂的sem图;
58.图7为对比例1及实施例1所得催化剂的tem图。
具体实施方式
59.下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
60.除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
61.实施例1
62.re/mo2ct
x
催化剂的制备
63.(1)使用60ml浓度不低于40wt%的氢氟酸刻蚀2g mo2ga2c前驱体材料,在55℃下搅
拌5天得到悬浊液,再将悬浊液用去离子水洗涤,使用滤膜过滤至滤液ph为7后,-50℃冷冻干燥8h,得到黑色粉末;
64.(2)将步骤(1)得到的0.8g黑色粉末用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合液分散,超声30min,用滤膜过滤后,分别用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合碱液、去离子水洗涤,冷冻干燥后得到mo2ct
x
;
65.(3)然后再将步骤(2)得到的mo2ct
x
负载高铼酸铵(nh4reo4)进行调控,使用初湿浸渍法,具体为:将0.3g mo2ct
x
浸渍在1ml nh4reo4溶液中,其中nh4reo4的浓度为0.0436g/ml,30℃下浸渍24h,干燥后置于管式炉中,在40-60ml/min的10vol%h2/ar气氛下,以2℃/min的升温速率,在400℃下焙烧4h,最终得到re/mo2ct
x
催化剂。经测试,re的负载量为10wt%,且以纳米颗粒形式存在。
66.实施例2
67.fe/mo2ct
x
催化剂的制备
68.(1)使用60ml浓度不低于40wt%的氢氟酸刻蚀2g mo2ga2c前驱体材料,在55℃下搅拌5天得到悬浊液,再将悬浊液用去离子水洗涤,使用滤膜过滤至ph为7后,-50℃冷冻干燥8h,得到黑色粉末;
69.(2)将步骤(1)得到的0.8g黑色粉末用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合液分散,超声30min,用滤膜过滤后,分别用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合碱液、去离子水洗涤,冷冻干燥后得到mo2ct
x
;
70.(3)然后再将步骤(2)得到的mo2ct
x
负载硝酸铁(fe(no3)3·
9h2o)进行调控,使用初湿浸渍法,将0.3g mo2ct
x
浸渍在1ml fe(no3)3·
9h2o溶液中,其中fe(no3)3·
9h2o的浓度为0.2204g/ml,30℃下浸渍24h,干燥后置于管式炉中,在40-60ml/min的10vol%h2/ar气氛下,以2℃/min的升温速率,在400℃下焙烧4h,最终得到fe/mo2ct
x
催化剂。经测试,fe的负载量为10wt%,且以纳米颗粒形式存在。
71.实施例3
72.co/mo2ct
x
催化剂的制备
73.(1)使用60ml浓度不低于40wt%的氢氟酸刻蚀2g mo2ga2c前驱体材料,在55℃下搅拌5天得到悬浊液,再将悬浊液用去离子水洗涤,使用滤膜过滤至ph为7后,-50℃冷冻干燥8h得到黑色粉末;
74.(2)将步骤(1)得到的0.8g黑色粉末用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合液分散,超声30min,用滤膜过滤后,分别用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合碱液和去离子水洗涤,冷冻干燥后得到mo2ct
x
催化剂;
75.(3)然后再将(2)得到的mo2ct
x
催化剂负载硝酸钴(co(no3)2·
6h2o)进行调控,使用初湿浸渍法,将0.3g mo2ct
x
浸渍在1ml co(no3)2·
6h2o溶液中,其中co(no3)2·
6h2o的浓度为0.1500g/ml,30℃下浸渍24h,干燥后置于管式炉中,在40-60ml/min的10vol%h2/ar气氛下,以2℃/min的升温速率,在400℃下焙烧4h,最终得到co/mo2ct
x
催化剂。经测试,co的负载量为10wt%,且以纳米颗粒形式存在。
76.实施例4
77.ni/mo2ct
x
催化剂的制备
78.(1)使用60ml浓度不低于40wt%的氢氟酸刻蚀2g mo2ga2c前驱体材料,在55℃下搅拌5天得到悬浊液,再将悬浊液用去离子水洗涤,使用滤膜过滤至ph为7后,-50℃冷冻干燥8h得到黑色粉末;
79.(2)将步骤(1)得到的0.8g黑色粉末用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合液分散,超声30min,用滤膜过滤后,分别用总体积为1l的0.2mol/l氢氧化钠溶液和0.5mol/l氨水溶液的混合碱液和去离子水洗涤,冷冻干燥后得到mo2ct
x
催化剂;
80.(3)然后再将(2)得到的mo2ct
x
催化剂负载硝酸镍(ni(no3)2·
6h2o)进行调控,使用初湿浸渍法,将0.3g mo2ct
x
浸渍在1ml ni(no3)2·
6h2o溶液中,其中ni(no3)2·
6h2o的浓度为0.1520g/ml,30℃下浸渍24h,干燥后置于管式炉中,在40-60ml/min的10vol%h2/ar气氛下,以2℃/min的升温速率,在400℃下焙烧4h,最终得到ni/mo2ct
x
催化剂。经测试,ni的负载量为10wt%,且以纳米颗粒形式存在。
81.对比例1
82.mo2ct
x
催化剂的制备:由实施例1步骤(1)-(2)制备得到。
83.测试例1
84.图6为对比例1(图a)及实施例1(图b)所得催化剂的sem图,从图6可以看到催化剂均呈薄层状结构。
85.图7为对比例1(图a)及实施例1(图b)所得催化剂的tem图,从图b中可以看到,re在mo2ct
x
上以纳米颗粒形式存在,纳米颗粒的平均尺寸为3.2
±
1.1nm。
86.测试例2、氨合成催化剂性能评价
87.取对比例1和实施例1-4的催化剂各0.20g,质量空速为60,000ml g-1
h-1
,在氨合成催化剂性能评价装置上进行氨合成速率测定,出口尾气中nh3浓度变化通过离子色谱(thermo scientific,dionex,ics-600)测定,反应气体组成为:25vol%n2 75vol%h2。
88.图1为对比例1及实施例1-4所得催化剂的合成氨性能,测试条件为:400℃、1mpa下进行氨合成反应。从图1可以看出,在400℃和1mpa下,实施例1中re/mo2ct
x
的合成氨性能最好,为20.5mmol g
cat-1
h-1
,是对比例1中mo2ct
x
的2倍。
89.图2为对比例1和实施例1所得催化剂在350℃,1mpa下进行氨合成反应的n2反应级数结果曲线。从图2可以看出,re/mo2ct
x
催化剂的n2反应级数为0.18,mo2ct
x
催化剂的n2反应级数为0.26,表明本发明的催化剂对于n2具有较强活化能力。
90.图3为对比例1和实施例1所得催化剂在1mpa和不同温度下进行氨合成反应的氨合成速率结果曲线。图3结果表明,本发明实施例1中re/mo2ct
x
催化剂的活化能为56.5
±
3kj/mol,而对比例1mo2ct
x
的活化能为83.1
±
3kj/mol,本发明较低的活化能表明本发明制备的催化剂可能不遵循解离路径,n≡n的解离不再是反应的决速步骤。
91.图4为对比例1和实施例1所得催化剂在400℃、1mpa下的热稳定性测试结果曲线。可以看出,re/mo2ct
x
和mo2ct
x
在400℃、1mpa下反应200h后依然较稳定,没有明显的失活,表明其呈现较高的热稳定性。
92.测试例3
93.甲烷化实验的测试条件:取催化剂0.20g,反应条件为400℃、1mpa,质量空速为60,000ml g-1
h-1
,在氨合成催化剂性能评价装置上进行氨合成反应后,出口尾气中测试甲烷浓
度,甲烷浓度利用气相色谱(gc-9560)进行测试,反应气体组成为:25vol%n2 75vol%h2。
94.图5为对比例1所得催化剂在400℃,1mpa下的甲烷化实验测试结果图,从图5可以看出,经长周期运行后无甲烷化现象,呈现极强的催化稳定性。由此确定,各实施例所得催化剂同样具有极强的催化稳定性。
95.以上,对本发明的实施方式进行了示例性的说明。但是,本发明的保护范围不拘囿于上述实施方式。凡在本发明的精神和原则之内,本领域技术人员所作出的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
