1.本发明涉及一种包含改性乙烯醇系聚合物的聚乙烯醇系树脂组合物。此外,本发明涉及一种悬浮聚合用分散稳定剂,尤其是适于乙烯基系化合物、特别是氯乙烯的悬浮聚合的分散稳定剂。
背景技术:
2.在使氯乙烯单体、或氯乙烯单体和能与其共聚的单体的混合物进行悬浮聚合的情况下,需要使用各种分散稳定剂,可使用聚乙烯醇、羟甲基纤维素等分散稳定剂,其中聚乙烯醇(pva)具有优异的性质,通常最常使用。例如,作为乙烯基系化合物的悬浮聚合用分散稳定剂,提出了:在乙烯醇系聚合物的末端导入源自醛的羰基,通过在皂化时受到脱水反应或脱乙酸反应而导入不饱和双键(例如参照专利文献1);使用在侧链具有特定的氧亚烷基的改性pva的方法等(例如参照专利文献2~3)。
3.现有技术文献
4.专利文献
5.专利文献1:日本特开平8-208724号公报
6.专利文献2:国际公开第2010/113569号
7.专利文献3:国际公开第2013/115239号
技术实现要素:
8.发明所要解决的问题
9.在这些方法中,近年来使用的大型聚合罐等各种类型的聚合罐无法充分应对。即,成为缺乏分散力且增塑剂吸收性低的乙烯基系树脂粒子,或者得到虽然分散力强但缺乏保护胶体性且粗大化的乙烯基系树脂粒子而使加工性降低,或者虽然为微细的乙烯基系树脂粒子但体积比重(bulk specific gravity)低等,在稳定地得到满意的乙烯基系树脂粒子方面不充分。
10.鉴于上述事实,在一个实施方式中,本发明的技术问题之一在于,提供一种聚乙烯醇系树脂组合物,其在将氯乙烯那样的乙烯基系化合物悬浮聚合时,作为适合得到微细且粒度的均匀性高、体积比重高、增塑剂吸收性适当的树脂粒子的分散稳定剂是有用的。
11.用于解决问题的方案
12.本发明人等为了解决上述问题而反复进行了深入研究,结果发现:使用如下聚乙烯醇系树脂组合物是有效的,所述聚乙烯醇系树脂组合物包含具有规定的聚氧亚烷基单元(以下,称为“亚烷基改性基团”)的改性乙烯醇系聚合物(a)、以及在聚乙烯醇链具有双键的改性乙烯醇系聚合物(b)。
13.因此,在一个方案中,本发明为聚乙烯醇系树脂组合物,其包含:通式(i)所示的聚氧亚烷基单元与聚乙烯醇链键合而成的改性乙烯醇系聚合物(a)、以及在聚乙烯醇链具有
双键的改性乙烯醇系聚合物(b)。
[0014][0015]
(式中,r1和r2分别独立地为甲基或乙基或氢原子,r3为烷基或氢原子。n表示重复单元数量,是5≤n≤70的整数。)
[0016]
在本发明的一个方案的聚乙烯醇系树脂组合物的一个实施方式中,所述改性乙烯醇系聚合物(a)与所述改性乙烯醇系聚合物(b)的重量比(a/b)为15/85~85/15。
[0017]
在本发明的一个方案的聚乙烯醇系树脂组合物的另一个实施方式中,所述改性乙烯醇系聚合物(b)的0.2重量%水溶液的、光程长1cm、波长280nm时的uv吸光度(abs)为0.1以上且3.0以下。
[0018]
在本发明的聚乙烯醇系树脂组合物的又一个实施方式中,所述改性乙烯醇系聚合物(a)的聚氧亚烷基单元的改性率为0.01~5摩尔%。
[0019]
在另一个方案中,本发明为悬浮聚合用分散稳定剂,其含有本发明的聚乙烯醇系树脂组合物。
[0020]
在又一个方案中,本发明为乙烯基系树脂的制造方法,其包括:使用本发明的悬浮聚合用分散稳定剂,使乙烯基系化合物单体、或乙烯基系化合物单体和能与所述乙烯基系化合物单体共聚的单体的混合物分散于水中而进行悬浮聚合的步骤。
[0021]
发明效果
[0022]
在使用本发明的悬浮聚合用分散稳定剂进行乙烯基系化合物的悬浮聚合的情况下,粗大粒子的形成少,可得到粒径的均匀性高的树脂粒子。由于粗大粒子的形成少,因此聚合时的成块得到抑制,得到粒径的均匀性高的粒子,因此垢(scale)附着减少。此外,得到高体积比重的树脂,树脂加工时的生产率提高。而且,也能得到增塑剂吸收性适当的树脂粒子。如此,本发明的悬浮聚合用分散稳定剂能兼备现有技术难以实现的要求性能。
具体实施方式
[0023]
本发明的悬浮聚合用分散稳定剂在一个实施方式中,包含:通式(i)所示的聚氧亚烷基单元与聚乙烯醇链键合而成的改性乙烯醇系聚合物(a)、以及在聚乙烯醇链具有双键的改性乙烯醇系聚合物(b)。需要说明的是,以下的说明中的“改性pva”是指改性乙烯醇系聚合物。
[0024][0025]
(式中,r1和r2分别独立地为甲基或乙基或氢原子,r3为烷基或氢原子。n表示重复
单元数量,是5≤n≤70的整数。)
[0026]
通式(i)中,n优选为5以上,更优选为10以上,更进一步优选为15以上,最优选为20以上。此外,n优选为70以下,更优选为60以下。
[0027]
表示r3的烷基例如可以为碳原子数1~10的烷基,优选为碳原子数1~5的烷基,更优选为碳原子数1~3的烷基。作为表示r3的烷基的具体例子,可列举出:甲基、乙基、正丙基、异丙基。
[0028]
从防止因ph的变化而脱离、无法发挥性能的观点考虑,通式(i)所示的部分优选仅经由醚键和/或碳-碳键与聚乙烯醇链键合。
[0029]
在一个实施方式中,在聚乙烯醇链具有双键的改性乙烯醇系聚合物(b)在聚乙烯醇链内具有通式(ii)所示的结构。
[0030][0031]
(式中,n表示重复单元数量,是1≤n≤10的整数。)
[0032]
通式(ii)中,n优选为1以上,更优选为2以上。此外,n优选为10以下,更优选为5以下。
[0033]
本发明的聚乙烯醇系树脂组合物优选所述改性乙烯醇系聚合物(a)与所述改性乙烯醇系聚合物(b)的重量比(a/b)为15/85~85/15。若所述重量比(a/b)为15/85以下,则未发挥分散力,其结果是,使乙烯基系树脂的增塑剂吸收性降低。此外,若重量比(a/b)为85/15以上,则有起因于改性乙烯醇系聚合物(b)的保护胶体性不足、乙烯基系树脂粗大化的隐患。因此,所述改性乙烯醇系聚合物(a)与所述改性乙烯醇系聚合物(b)的重量比(a/b)优选为15/85~85/15,更优选为20/80~80/20,更进一步优选为25/75。需要说明的是,在本发明中,通式(i)所示的聚氧亚烷基单元与聚乙烯醇链键合、并且在聚乙烯醇链具有双键的改性乙烯醇系聚合物视为既不相当于改性乙烯醇系聚合物(a)也不相当于改性乙烯醇系聚合物(b)。
[0034]
所述改性乙烯醇系聚合物(b)优选0.2重量%水溶液的、光程长1cm、波长280nm时的uv吸光度(abs)为0.1以上且3.0以下。该波长280nm的吸收相当于共轭双键的双链,若吸光度小于0.1,则双键不足,保护胶体性降低,因此,其结果是,不易得到具有适度的粒径的乙烯基系树脂。此外,若吸光度为3.0以上,则着色变得显著,在用作分散剂时,对乙烯基系树脂的着色造成影响。此外,有时化学上变得不稳定,水溶液的粘度变高,或凝胶化。因此,改性乙烯醇系聚合物(b)的0.2重量%水溶液的波长280nm时的uv吸光度需要为0.1以上且3.0以下,优选为0.2以上且2.5以下,进一步优选为0.3以上且2.0以下。
[0035]
所述改性乙烯醇系聚合物(a)也取决于通式(i)所示的聚氧亚烷基单元的种类,但所述亚烷基单元的改性率(以下,也称为“亚烷基改性率”)优选为0.01摩尔%以上且5摩尔%以下。若亚烷基改性率超过5摩尔%,则无法保持每一分子改性pva(a)所含的亲水基团/疏水基团的平衡,有时虽然分散力强但缺乏保护胶体性,在该情况下,难以用作悬浮聚合用分散稳定剂。由此,亚烷基改性率优选为5摩尔%以下,更优选为4摩尔%以下,更进一
步优选为2摩尔%以下。另一方面,在亚烷基改性率小于0.01摩尔%的情况下,该改性pva(a)中所含的改性基团的数量少,未充分体现要求物性。由此,亚烷基改性率为0.01摩尔%以上是重要的,优选为0.05摩尔%以上,更优选为0.1摩尔%以上。
[0036]
亚烷基改性率是指相对于构成改性pva(a)的聚乙烯醇链的全部单体单元的摩尔数的、与具有上述通式(i)所示的部分的聚氧亚烷基单元键合的所述单体单元的摩尔数的比例(摩尔%)。亚烷基改性率可以利用质子nmr求出。具体而言,将改性pva皂化至皂化度99.95摩尔%以上后,充分进行甲醇清洗,制作分析用的改性pva。将制作出的分析用的改性pva溶解于重水,进一步加入几滴naoh重水溶液成为ph=14后,使用质子nmr在80℃下进行测定。在根据氧亚乙基部分(例:r1=h、r2=h)计算的情况下,根据归属于改性pva的聚乙烯醇链的亚甲基的1.2~1.8ppm的峰的积分值和归属于氧亚乙基部分的3.6~3.7ppm的峰的积分值,通过常规方法计算出含量。具体而言,当将改性pva的聚乙烯醇链的亚甲基的积分值设为b,将氧亚乙基部分的积分值设为a,将氧亚乙基部分的重复单元数量设为x时,鉴于质子数(亚甲基为2h,亚乙基为4h),亚烷基改性率计算为{a/(4
×
x)}/(b/2)
×
100(mol%)。例如,在a=1,x=1,b=100的情况下,计算为0.5mol%。此外,在根据氧亚丁基或氧亚丙基部分计算的情况下,根据归属于改性pva的聚乙烯醇链的亚甲基的1.2~1.8ppm的峰的积分值和归属于氧亚丁基部分(r1=h,r2=ch2ch3(或r1=ch2ch3,r2=h))或氧亚丙基部分(r1=h,r2=ch3(或r1=ch3,r2=h))的末端甲基的0.80~0.95ppm的峰的积分值,通过常规方法计算出含量。具体而言,当将改性pva的聚乙烯醇链的亚甲基的积分值设为b,将氧亚丁基部分或氧亚丙基部分的积分值设为c,将重复单元数量设为y时,鉴于质子数(亚甲基为2h,甲基为3h),亚烷基改性率计算为{c/(3
×
y)}/(b/2)
×
100(mol%)。例如,在c=1、y=1、b=100的情况下,计算为0.67mol%。需要说明的是,在改性pva具有氧亚乙基部分、以及氧亚丙基部分或氧亚丁基部分这双方的情况下,与根据氧亚乙基部分计算出的亚烷基改性率相比,基于归属于氧亚丙基部分或氧亚丁基部分的末端甲基的峰的积分值计算出的亚烷基改性率的测定精度更高,因此在两者的值不同的情况下,采用基于归属于氧亚丙基部分或氧亚丁基部分的末端甲基的峰的积分值计算出的亚烷基改性率。
[0037]
为了提高使乙烯基系化合物进行悬浮聚合时的分散稳定性,所述改性乙烯醇系聚合物(a)的粘度平均聚合度优选为400以上,更优选为500以上。此外,为了不使分散力降低,改性pva的粘度平均聚合度优选为3000以下,更优选为2000以下,更进一步优选为1500以下,更进一步优选为1000以下。
[0038]
为了提高使乙烯基系化合物进行悬浮聚合时的分散稳定性,所述改性乙烯醇系聚合物(b)的粘度平均聚合度优选为600以上,更优选为700以上。此外,为了不使分散力降低,改性pva的粘度平均聚合度优选为4000以下,更优选为3000以下,更进一步优选为2000以下,更进一步优选为1500以下。
[0039]
粘度平均聚合度依据jis k6726:1994测定。即,将改性pva完全皂化,进行纯化后,根据在30℃的水或二甲基亚砜(dmso)中测定出的极限粘度[η]求出。
[0040]
从水溶性的观点考虑,所述改性乙烯醇系聚合物(a)的皂化度需要为65摩尔%以上。此外,为了提高使乙烯基系化合物进行悬浮聚合时所得到的粒子的孔隙率(porosity)而提高增塑剂吸收性,改性pva的皂化度优选为99.9摩尔%以下,更优选为90摩尔%以下,更进一步优选为80摩尔%以下。
[0041]
从水溶性的观点考虑,所述改性乙烯醇系聚合物(b)的皂化度需要为65摩尔%以上。此外,为了提高使乙烯基系化合物进行悬浮聚合时所得到的粒子的孔隙率而提高增塑剂吸收性,改性pva的皂化度优选为99.9摩尔%以下,更优选为90摩尔%以下,更进一步优选为80摩尔%以下。
[0042]
改性pva的皂化度依据jis k6726:1994测定。即,可以利用氢氧化钠对试样中的残存乙酸基(摩尔%)进行定量,从100减去,由此求出。
[0043]
所述改性乙烯醇系聚合物(a)的制造方法没有特别限制,可以使用使pva的羟基与聚氧亚烷基和有机酸反应,进行接枝的方法等,但如下方法是容易且经济的,因此优选使用,即,在有机酸的共存下,将乙酸乙烯酯所代表的乙烯酯系单体与具有通式(i)所示的聚氧亚烷基单元的不饱和单体共聚而得到改性乙烯酯系聚合物的工序;以及对所得到的改性乙烯酯系聚合物进行皂化。
[0044]
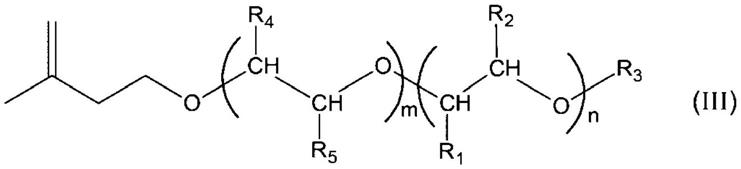
作为衍生出通式(i)所示的改性结构的不饱和单体,可列举出:聚氧亚烷基烯基醚、聚氧亚烷基单(甲基)烯丙基醚、聚氧亚烷基单乙烯基醚等,具体而言,可列举出:聚氧亚丁基聚氧亚乙基烯基醚、聚氧亚丙基聚氧亚乙基烯基醚、聚氧亚丁基烯基醚、聚氧亚丁基聚氧亚丙基烯基醚、聚氧亚丙基烯基醚、聚氧亚丙基聚氧亚乙基烯基醚、聚氧亚丁基聚氧亚乙基单烯丙基醚、聚氧亚丙基聚氧亚乙基单烯丙基醚、聚氧亚丁基单烯丙基醚、聚氧亚丁基聚氧亚丙基单烯丙基醚、聚氧亚丙基单烯丙基醚、聚氧亚丙基聚氧亚乙基单烯丙基醚、聚氧亚丁基聚氧亚乙基单乙烯基醚、聚氧亚丙基聚氧亚乙基单乙烯基醚、聚氧亚丁基单乙烯基醚、聚氧亚丁基聚氧亚丙基单乙烯基醚、聚氧亚丙基单乙烯基醚、聚氧亚丙基聚氧亚乙基单乙烯基醚等。其中,从反应性、性能的方面考虑,进一步优选使用如下述的通式(iii)所示的醚。作为如通式(iii)所示的聚氧亚烷基烯基醚的具体例子,可列举出聚氧亚丁基聚氧亚乙基烯基醚。
[0045][0046]
(通式(iii)中,r1和r2分别独立地为甲基或乙基或氢原子,r3为烷基或氢原子。n表示重复单元数量,是5≤n≤70的整数。r4和r5中一方为甲基或乙基,另一方为氢原子。m表示重复单元数量,是1≤m≤30的整数。其中,重复单元数量n的部分与重复单元数量m的部分不同。)
[0047]
作为乙烯酯系单体,除了乙酸乙烯酯以外,可列举出:甲酸乙烯酯、丙酸乙烯酯、戊酸乙烯酯、癸酸乙烯酯、月桂酸乙烯酯、硬脂酸乙烯酯、苯甲酸乙烯酯、新戊酸乙烯酯以及叔碳酸(versatic acid)乙烯酯等。
[0048]
作为在所述改性乙烯醇系聚合物(b)导入如通式(ii)所示的双键的方法,没有特别限定,可列举出周知的聚合方法,例如利用过氧化氢、过碘酸等氧化剂将未改性pva系树脂进行氧化处理并进行热处理的方法;例如,在含有羰基的醛、酮等链转移剂、不饱和羧酸那样的共聚单体的共存下进行聚合,将所得到的聚乙酸乙烯酯皂化,引发脱水反应而导入的方法;一边吹入氧一边进行聚合,将所得到的聚乙酸乙烯酯进行皂化,引发脱水反应而导
入的方法。在工业上,下述方法特别有利:在含有羰基的醛、酮等链转移剂、不饱和羧酸那样的共聚单体的共存下进行聚合,将所得到的聚乙酸乙烯酯皂化,引发脱水反应而得到在聚乙烯醇链具有碳-碳双键的改性pva系树脂。
[0049]
作为用于对所述改性乙烯醇系聚合物(b)衍生双键的改性种,可列举出:能与乙烯酯系单体共聚的马来酸二甲酯、马来酸单甲酯等不饱和羧酸烷基酯,乙醛、丙醛、正丁醛等烷基醛,四氯化碳、溴丁烷等卤素系烃。
[0050]
作为进行该共聚而采用的聚合方式,可以为间歇聚合、半间歇聚合、连续聚合、半连续聚合中的任意方式。作为聚合方法,可以从本体聚合法、溶液聚合法、悬浮聚合法、乳液聚合法等公知的方法之中采用任意的方法。其中,所使用的改性种大多具有对聚合粒径造成影响的水溶性、表面活性能力,因此不采用需要控制聚合粒径的悬浮聚合和乳液聚合,而优选采用在醇系溶剂存在下进行聚合的溶液聚合法或在不使用溶剂的情况下进行聚合的本体聚合法。作为用于溶液聚合法的醇系溶剂,可以使用甲醇、乙醇、异丙醇等,但并不限定于此。此外,这些溶剂可以单独使用,也可以并用两种以上。
[0051]
使乙烯酯系单体进行自由基聚合时的聚合引发剂没有特别限定,可以单独或组合两种以上使用:偶氮双异丁腈、偶氮双-2,4-二甲基戊腈、偶氮双(4-甲氧基-2,4-二甲基戊腈)、偶氮双二甲基戊腈、偶氮双甲氧基戊腈等偶氮化合物;过氧化乙酰、过氧化苯甲酰、过氧化月桂酰、过氧化乙酰基环己基磺酰、2,4,4-三甲基戊基-2-过氧化苯氧基乙酸酯等过氧化物;过氧化二碳酸二异丙酯、过氧化二碳酸二-2-乙基己酯、过氧化二碳酸二乙氧基乙酯等过碳酸酯化合物;过氧化新癸酸叔丁酯、过氧化新癸酸α-异丙苯酯等过酸酯(perester)化合物等。
[0052]
此外,在高的温度下进行共聚的情况下,有时观察到因乙烯酯系单体的分解而导致的pva的着色等。在该情况下,出于防止着色的目的,向聚合体系中添加1ppm以上且100ppm以下(相对于乙烯酯系单体的质量)左右的柠檬酸那样的抗氧化剂也没有什么影响。
[0053]
制造本发明的改性pva时的皂化方法也没有特别限定,优选对通过上述的方法得到的聚合物按照常规方法,以兼用作溶剂的方式来使用醇类。作为醇,可列举出:甲醇、乙醇、丁醇等。醇中的聚合物的浓度可以选自20~50质量%的范围。作为碱催化剂,可以使用氢氧化钠、氢氧化钾、甲醇钠、乙醇钠、甲醇钾等碱金属的氢氧化物、醇化物这样的碱催化剂,作为酸催化剂,可以使用盐酸、硫酸等无机酸水溶液、对甲苯磺酸等有机酸。这些催化剂的使用量相对于乙烯酯系单体需要设为1~100毫摩尔当量。皂化温度没有特别限制,理想的是,通常从10~70℃的范围选择,优选从30~50℃的范围选择。反应通常进行0.5~3小时。
[0054]
本发明的悬浮聚合用分散稳定剂也可以在不损害本发明的主旨的范围内,含有上述改性pva以外的pva、其他各种添加剂。作为该添加剂,例如,可列举出醛类、卤代烃类、硫醇类等聚合调节剂;酚化合物、硫化合物、n-氧化物化合物等聚合抑制剂;ph调整剂;交联剂;防腐剂;防霉剂、防粘连剂;消泡剂等。从显著地发挥本发明的效果的观点考虑,本发明的悬浮聚合用分散稳定剂优选含有合计10质量%以上、更优选含有30质量%以上、更进一步优选含有70质量%以上的改性pva(a)和改性pva(b)。
[0055]
本发明的悬浮聚合用分散稳定剂特别是能优选用于乙烯基系化合物的悬浮聚合。因此,根据本发明的另一个方案,提供一种乙烯基系树脂的制造方法,其包括:使用悬浮聚
合用分散稳定剂,使乙烯基系化合物单体、或乙烯基系化合物单体和能与其共聚的单体的混合物分散于水中而进行悬浮聚合的步骤。
[0056]
作为乙烯基系化合物,可列举出:氯乙烯等卤代乙烯;乙酸乙烯酯、丙酸乙烯酯等乙烯酯;丙烯酸、甲基丙烯酸、它们的酯和盐;马来酸、富马酸、它们的酯和酸酐;苯乙烯、丙烯腈、偏氯乙烯、乙烯醚等。其中,本发明的一个实施方式的悬浮聚合用分散稳定剂特别优选用于使氯乙烯单独进行悬浮聚合时,或者使氯乙烯和能与氯乙烯共聚的单体一起进行悬浮聚合时。作为能与氯乙烯共聚的单体,可列举出:乙酸乙烯酯、丙酸乙烯酯等乙烯酯;(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯等(甲基)丙烯酸酯;乙烯、丙烯等α-烯烃;马来酸酐、衣康酸等不饱和二羧酸类;丙烯腈、苯乙烯、偏氯乙烯、乙烯基醚等。
[0057]
本发明的悬浮聚合用分散稳定剂可以单独使用或也可以与其他稳定剂例如纤维素系衍生物、表面活性剂等并用。
[0058]
通过使用本发明的悬浮聚合用分散稳定剂,可得到树脂粒子的体积比重高、粒度分布均匀且物性非常优异的氯乙烯树脂。以下,对乙烯基系化合物的聚合法举例进行具体说明,但并不限定于此。
[0059]
在制造氯乙烯树脂粒子等乙烯基系化合物的树脂粒子的情况下,相对于乙烯基系化合物单体,添加0.01质量%~0.3质量%,优选添加0.04质量%~0.15质量%的上述的悬浮聚合用分散稳定剂。此外,乙烯基系化合物与水之比按质量比计可以设为乙烯基系化合物:水=1∶0.9~1∶3,优选为乙烯基系化合物:水=1∶1~1∶1.5。
[0060]
聚合引发剂为以往用于乙烯基系化合物的聚合的聚合引发剂即可,该聚合引发剂可以单独或组合使用:过氧化二碳酸二异丙酯、过氧化二碳酸二-2-乙基己酯、过氧化二碳酸二乙氧基乙酯等过碳酸酯化合物;过氧化新癸酸叔丁酯、过氧化新癸酸α-异丙苯酯等过酸酯化合物;过氧化乙酰基环己基磺酰、2,4,4-三甲基戊基-2-过氧化苯氧基乙酸酯等过氧化物;偶氮双-2,4-二甲基戊腈、偶氮双(4-甲氧基-2,4-二甲基戊腈)等偶氮化合物;进而过硫酸钾、过硫酸铵、过氧化氢等。
[0061]
而且,也可以任意添加适合用于乙烯基系化合物的聚合的聚合调节剂、链转移剂、凝胶化改良剂、防静电剂、ph调整剂等。
[0062]
实施乙烯基系化合物的聚合时的各成分的装料比例、聚合温度等依照以往在乙烯基系化合物的悬浮聚合中采用的条件来确定即可,不存在特别限定的理由。
[0063]
实施例
[0064]
以下,列举出实施例,对本发明进一步详细地进行说明。
[0065]
(pva1的制造)
[0066]
向聚合罐中装入乙酸乙烯酯1850g、甲醇1000g、作为改性种的通式(iii)所示的m=5~9、n=45~55的聚氧亚烷基烯基醚(单体a)133g,在体系内进行了30分钟氮置换。关于单体a,通过nmr确认到m=5~9,n=45~55。向聚合罐中装入偶氮双异丁腈0.25g,在60℃下进行了9小时聚合后,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进行皂化而制作pva1。通过上述的分析法对所得到的pva1的粘度平均聚合度、皂化度以及改性率进行了测定,其结果是,粘度平均聚合度为750,皂化度为73摩尔%,改性率为0.18摩尔%。
[0067]
使用紫外可见近红外分光光度计((株)岛津制作所制分光光度计uv1800),对光程
长1cm、波长280nm时的、聚乙烯醇系分散剂的0.2重量%水溶液的吸光度进行了测定,其结果是0.02abs。需要说明的是,使用了厚度1cm的试样容器(池:cell)。
[0068]
(pva2的制造)
[0069]
向聚合罐中装入乙酸乙烯酯1800g、甲醇612g、马来酸二甲酯4g,在体系内进行了30分钟氮置换。向聚合罐中装入偶氮双异丁腈0.3g,一边滴加乙酸乙烯酯400g、马来酸二甲酯35g,一边在60℃下进行了11小时聚合后,停止滴加,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进行皂化而制作pva2。求出有机酸改性率的方法没有特别限定,可以利用酸价等求出,但利用质子nmr求出是简便的,因此在此利用质子nmr求出。具体而言,将改性pva完全皂化至皂化度99.95摩尔%以上后,充分进行甲醇清洗,制作分析用的改性pva。在使用马来酸二甲酯作为改性种的情况下,在完全皂化的过程中,羧基生成。因此,改性种的改性率与有机酸改性率相等。将制作出的分析用的改性pva溶解于重水,进一步加入几滴naoh重水溶液成为ph=14后,在80℃下测定,得到1h-nmr光谱。以改性pva的聚乙烯醇链的亚甲基(1.2~1.8ppm)的峰的积分值作为基准计算出有机酸改性率。例如,在有机酸单元具有羧基的情况下,根据与羧基邻接的碳的氢原子的2.2~2.9ppm的峰计算出。若以有机酸单元具有羧基的情况为例,则当将改性pva的聚乙烯醇链的亚甲基的积分值设为b,将与羧基邻接的碳的氢原子的积分值设为a时,鉴于质子数(亚甲基为2h),改性率计算为a/(b/2)
×
100(mol%)。例如,在a=1、b=100的情况下,计算为2.0mol%。
[0070]
通过上述的分析法对pva2的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0071]
(pva3的制造)
[0072]
向聚合罐中装入乙酸乙烯酯1825g、甲醇1740g、马来酸二甲酯2g,在体系内进行了30分钟氮置换。向聚合罐中装入偶氮双异丁腈0.3g,一边滴加乙酸乙烯酯550g、马来酸二甲酯22g,一边在60℃下进行了11小时聚合后,停止滴加,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进行皂化而制作pva3。通过上述的分析法对pva3的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0073]
(pva4的制造)
[0074]
向聚合罐中装入乙酸乙烯酯1800g、甲醇612g、马来酸二甲酯7g,在体系内进行了30分钟氮置换。向聚合罐中装入偶氮双异丁腈0.3g,一边滴加乙酸乙烯酯400g、马来酸二甲酯63g,一边在60℃下进行了11小时聚合后,停止滴加,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进行皂化而制作pva4。通过上述的分析法对pva4的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0075]
(pva5的制造)
[0076]
向聚合罐中装入乙酸乙烯酯1800g、甲醇612g、马来酸二甲酯10g,在体系内进行了30分钟氮置换。向聚合罐中装入偶氮双异丁腈0.3g,一边滴加乙酸乙烯酯400g、马来酸二甲酯88g,一边在60℃下进行了11小时聚合后,停止滴加,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进
行皂化而制作pva5。通过上述的分析法对pva5的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0077]
(pva6的制造)
[0078]
向聚合罐中装入乙酸乙烯酯3000g、改性种的乙醛15g,在体系内进行了30分钟氮置换。向聚合罐中装入偶氮双异丁腈0.2g,在65~75℃下进行了6小时聚合后,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进行皂化,在120℃下使其干燥6小时而制造pva6。在使用乙醛作为改性种的情况的改性率与羰基改性率相等。羰基改性率利用质子nmr求出。具体而言,将改性pva完全皂化至皂化度99.95摩尔%以上后,充分进行甲醇清洗,制作分析用的改性pva。将制作出的分析用的改性pva溶解于重水,进一步加入几滴naoh重水溶液成为ph=14后,在80℃下测定,得到1h-nmr光谱。以改性pva的主链的亚甲基(1.2~1.8ppm)的峰的积分值为基准,在乙醛的情况下,利用甲基末端(2.15~2.35ppm)的峰的积分值计算出。具体而言,当将改性pva的主链的亚甲基的积分值设为b,将乙醛的甲基末端的积分值设为d时,鉴于质子数(亚甲基为2,乙醛的甲基末端为x=3),羰基改性率计算为(d/3)/(b/2)
×
100(mol%)。通过上述的分析法对pva6的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0079]
(pva7的制造)
[0080]
向聚合罐中装入乙酸乙烯酯1220g、甲醇1350g、作为改性种的通式(iii)所示的m=5~9、n=15~25的聚氧亚烷基烯基醚(单体b)52g,在体系内进行了30分钟氮置换。关于单体b,通过nmr确认到m=5~9,n=15~25。向聚合罐中装入偶氮双异丁腈0.25g,在60℃下进行了9小时聚合后,冷却而停止聚合。接着,通过常规方法去除未反应的乙酸乙烯酯,通过常规方法,利用氢氧化钠甲醇溶液对所得到的聚合物进行皂化而制作出pva7。通过上述的分析法对pva7的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0081]
(pva8的制造)
[0082]
向聚合罐中装入n=15~25的聚乙二醇烯丙基醚(单体c)(日油株式会社提供uniox pka-5005)56g来代替单体a,除此以外,与pva1同样地制作出pva8。需要说明的是,关于单体c,通过nmr确认到n=15~25。通过上述的分析法对pva8的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0083]
(pva9的制造)
[0084]
向聚合罐中装入通式(iii)所示的m=15~25、n=15~25的聚乙二醇聚丙二醇烯丙基醚(单体d)(日油株式会社提供unireave pka-5013)74g来代替单体a,除此以外,与实施例1同样地制作出pva9。关于单体d,通过nmr确认到m=15~25,n=15~25。通过上述的分析法对pva9的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0085]
(pva10、11)
[0086]
pva8和pva9分别使用了denka(株)制聚乙烯醇w-24n和b-24。通过上述的分析法对pva8、pva9的粘度平均聚合度、皂化度、改性率以及0.2重量%水溶液的波长280nm时的uv吸光度进行了测定。将结果示于表1。
[0087]
[表1]
[0088][0089]
(实施例1~10和比较例1~4)
[0090]
按表2所示的重量比率利用诺塔混合机(nauta mixer)(注册商标)将各pva混合,得到聚乙烯醇系树脂组合物。将这些组合物作为分散稳定剂,进行氯乙烯的悬浮聚合并进行评价。
[0091]
〈氯乙烯的悬浮聚合〉
[0092]
向具备搅拌器的容量30l的不锈钢制高压釜中在搅拌下装入30℃的水10.1kg、聚乙烯醇系树脂组合物5.8g、作为聚合引发剂的过氧化新癸酸叔丁酯4.6g、过氧化新癸酸α-异丙苯酯1g。将高压釜进行真空脱气后,加入氯乙烯单体7.2kg,在57℃下进行4小时聚合。
[0093]
〈氯乙烯树脂的评价〉
[0094]
通过以下的方法对所得到的氯乙烯树脂的平均粒径、粒度分布、增塑剂吸收量以及体积比重进行了评价。
[0095]
平均粒径的测定依据jis z8815:1994,使用60目(网眼250μm)、80目(网眼180μm)、100目(网眼150μm)、150目(网眼106μm)、200目(网眼75μm)的筛,将60目(网眼250μm)上的粒子量相对于总体量的比率作为粗粒量,将累积频率50%(质量基准)的粒径(d50)作为平均粒径,将累积频率80%(质量基准)的粒径(d80)与累积频率20%(质量基准)的粒径(d20)之差作为粒度分布。
[0096]
体积比重基于jis k6720-2:1999进行了测定。
[0097]
增塑剂吸收量按以下的步骤测定。在内径25mm、深度85mm的铝合金制容器的底部填充玻璃纤维,投入氯乙烯树脂10g。在其中加入增塑剂(邻苯二甲酸二辛酯,以下为dop)15ml,放置30分钟而使dop充分渗透至氯乙烯树脂。之后,在1500g的加速度下将过剩的dop离心分离,测定氯乙烯树脂10g所吸收的dop的质量,换算成每100质量份氯乙烯树脂的dop质量份(phr)。
[0098]
将结果示于表2。在比较例1、3、4中不使用在聚乙烯醇链具有双键的改性乙烯醇系聚合物(b),因此氯乙烯树脂粒子粗大化,粗粒量多,粒径的均匀性差。在不使用改性乙烯醇系聚合物(a)的比较例2中,成为增塑剂吸收性差的氯乙烯粒子。相对于此,可知:若使用实施例1~10所示的聚乙烯醇系树脂组合物,则氯乙烯树脂中粗大粒子的形成少,可得到粒径的均匀性高的粒子。此外,增塑剂吸收性优异,而且,聚合时的成块、垢附着减少,可得到脱单体性优异的聚合物粒子。由此,实施例1~10的分散稳定剂在工业上极为有利。
[0099]
[表2]
[0100]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。