1.本发明涉及一种用于烷氧基羰基化反应的固体多相催化剂及其制备方法与应用,属于多相催化技术领域。
背景技术:
2.均相催化体系是一类较为经典的反应体系,在温和的反应条件下具有较高的催化活性和目的产物选择性,但存在着催化剂同反应物料难于分离的问题。多相催化与均相催化相比具有一个明显的优点,即催化剂与反应物料容易分离,存在的主要问题是反应条件苛刻,反应活性相对较低等。因此,开发一系列兼具均相催化和多相催化优点的新型固载化多相催化剂,是科学研究的热点。近年来,多孔有机聚合物材料的设计合成逐渐成为多孔材料研究领域新的热点之一。与传统的无机微孔材料和金属有机框架材料(mofs)相比,有机微孔聚合物所具有的骨架由纯粹的有机分子构成,相互之间通过共价键连接,具有开放的孔道与优异的孔性质。更重要的是,由于有机化学合成方法的多样性,为有机分子网络的构建提供了丰富的合成路径和构建方式,可以通过目的性的引入功能化的有机分子使材料具有相应的性质,通过调节有机分子的结构可以调控材料的孔性质。除此以外,金属功能化的多孔有机聚合物材料可以将具有催化活性的金属活性单元定点地引入到多孔有机聚合物中,实现以单原子形式高分散于有机共聚物载体中,这样不仅有利于稳定金属活性位点,而且在一定程度上提高了金属的利用效率。
3.醇类和烯烃以及co经催化剂催化制备酯类化合物的反应称之为烷氧基羰基化,产物酯类在工业领域有非常广泛的应用,可以用作有机合成中间体以及制造香料,酯类也是一种优良溶剂,在制造纤维素醚、酯时可以用作溶剂,也可以作为多种天然树脂和合成树脂的溶剂。但是,现有的烷氧基羰基化反应体系均为均相体系,难以将催化剂与反应液分离。
4.综上所述,对于适用于实际工业应用的烷氧基羰基化反应,绿色高效可回收利用的非均相催化剂是该领域的主要研究方向。
技术实现要素:
5.为了解决上述问题,本发明的目的在于提供一种由有机配体聚合物负载金属活性组分的固体多相催化剂及其制备方法和应用。
6.为此,本发明提供一种应用于烷氧基羰基化反应的固体多相催化剂,其特征在于:所述有机配体聚合物是由含有乙烯基官能团化的膦配体单体和含乙烯基的酸性有机物单体经溶剂热共聚合生成的具有大比表面积和多级孔结构的聚合物,其中所述金属组分是金属pd、ru、ni、ir、rh、mo、fe或cu中的一种或几种,所述金属组分与所述有机配体聚合物骨架中的p原子形成配位键,高分散且稳定的存在于有机配体聚合物载体上。
7.在一个优选实施方案中,所述金属组分在所述固体多相催化剂总重量中占0.01-17.0%,优选0.2-5%。
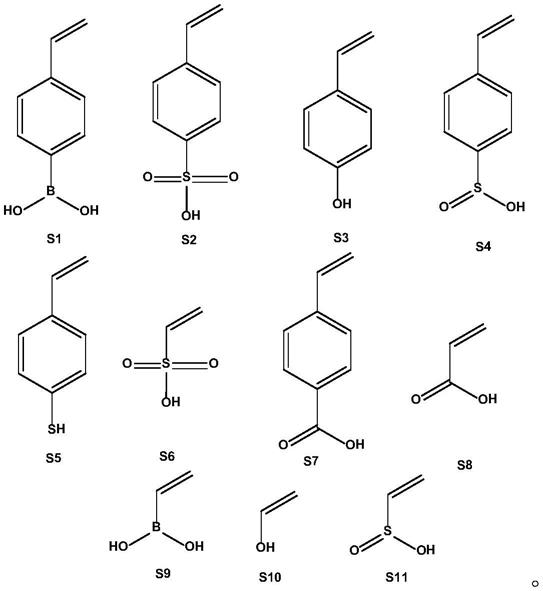
8.在一个优选实施方案中,所述含有乙烯基官能团化的膦配体,是选自以下各项中
的一种或几种:
[0009][0010]
在一个优选实施方案中,所述含乙烯基的酸性有机物单体,是选自以下各项中的一种或几种:
[0011][0012]
在一个优选实施方案中,所述有机配体聚合物的比表面积为100-3000m2/g,孔容
为0.1-5.0cm3/g,孔径分布在0.1-100.0nm。
[0013]
在一个优选实施方案中,所述固体多相催化剂的制备方法,包括制备步骤如下:a)在273-473k和惰性气体保护氛围下,在含有乙烯基官能团化的膦配体单体的溶剂中,加入自由基引发剂,再加入含乙烯基的酸性有机物单体,搅拌0.5-100小时;b)在323-473k和惰性气体保护氛围下,将步骤a)的溶液在水热高压釜中静置0.5-100小时,进行溶剂热聚合反应;c)将步骤b)结束后在273-473k温度条件下真空抽除溶剂,即得到所述有机配体聚合物;d)将所述有机配体聚合物置于含有活性金属组分的溶剂中,在273-473k和惰性气体保护氛围下搅拌0.5-100小时,然后在273-473k温度条件下真空抽除溶剂,即得到由有机配体聚合物负载活性金属组分的所述固体多相催化剂。
[0014]
在一个优选实施方案中,所述催化剂制备方法步骤a)和d)中使用的溶剂是苯、甲苯、四氢呋喃、甲醇、乙醇、二氯甲烷、二氯乙烷或去离子水中的一种或几种;步骤a)中使用的自由基引发剂是过氧化环己酮、过氧化二苯甲酰、叔丁基过氧化氢、偶氮二异丁腈或偶氮二异庚腈的一种或几种。
[0015]
在一个优选实施方案中,所述催化剂制备方法中所述自由基引发剂与所述有机单体的重量比为1:500-1:5。
[0016]
在一个优选实施方案中,所述的固体多相催化剂在烷氧基羰基化反应中的应用,是指在所述固体多相催化剂存在下使醇类原料与烯烃和co混合气在固定床、滴流床、浆态床或釜式反应器中进行所述烷氧基羰基化反应,其中反应温度为273-573k,反应压力为0.05-20.0mpa,醇类液时空速为0.01-20.0h-1
,烯烃和co气体空速为100-20000h-1
,所述烯烃原料与所述co原料的摩尔比为1:1-1:100。
[0017]
在一个优选实施方案中,所述有机配体聚合物合成时的惰性气体气氛为氩气、氦气以及氮气中的一种或二种以上。
[0018]
本发明产生的有益效果包括但不限于以下方面:
[0019]
本发明的固体多相催化剂与现有的烷氧基羰基化催化剂相比,催化剂制备方法简单;金属组分与聚合物载体中p原子之间因配位作用而稳定的存在于载体上;聚合物载体具有大比表面积和多级孔结构,金属组分可以高分散的存在于载体上,从而使本发明的固体多相催化剂具有优异的催化反应性能及较高的稳定性。此外,本发明的催化剂在宏观上是多相催化剂,因此在回收循环利用和与反应物及产物的分离等方面具有明显的优越性,具有广阔的工业应用前景。
[0020]
本发明催化剂在烷氧基羰基化反应中,一方面金属组分与聚合物载体中p原子之间因配位作用而稳定的存在于载体上;另一方面聚合物载体具有大比表面积和多级孔结构,金属组分可以高分散的存在于载体上,从而使本发明的固体多相催化剂具有优异的催化反应性能和稳定性,并且催化剂容易与反应物和产物分离,具有广阔工业应用前景。
附图说明
[0021]
图1是本发明的固体多相聚合物合成路线示意图。
[0022]
图2是本发明的固体多相催化剂的n2吸脱附等温曲线和孔径分布曲线图。
具体实施方式
[0023]
为了更好的说明本发明的催化剂的制备方法及其在烷氧基羰基化反应中的应用,下面举出一些催化剂样品的制备及其在反应工艺中的应用的实施例,但本发明不限于所列举的实例。除非另有具体说明,本技术中所用的“百分比”基于重量。
[0024]
实施例1
[0025]
在298k和氩气保护氛围下,将8.0克三(4-乙烯基苯)基膦溶于100ml四氢呋喃溶剂中,向上述溶液中加入0.25克自由基引发剂偶氮二异丁腈,再加入2.0g对乙烯基苯磺酸,搅拌0.5小时。将搅拌好的溶液移至水热高压釜中,于373k和氩气气体保护氛围下溶剂热法聚合24h。待上述聚合后冷却至室温,333k温度条件下真空抽除溶剂,即得到对应的的多孔有机聚合物。
[0026]
在298k和惰性气体保护氛围下,称取0.0114醋酸钯(ⅱ)溶于50ml四氢呋喃溶剂中,加入1.0克上述制备的多孔有机聚合物,搅拌24小时。随后,333k温度条件下真空抽除溶剂,即获得由有机配体聚合物负载金属组分的固体多相催化剂。本发明的固体多相催化剂合成路线示意图见图1,本发明的固体多相催化剂的n2吸脱附等温曲线和孔径分布曲线图见图2,并由此结果得到此催化剂具有不错的比表面积和丰富的多级孔道结构。金属负载量0.5wt%。
[0027]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入乙烯和co混合气(乙烯:co=1:2,体积比,下同),甲醇原料溶液经高压计量泵泵入反应器中开始反应,乙烯和甲醇制丙酸甲酯反应温度105℃,反应压力2mpa,甲醇液时空速0.1h-1
,co/甲醇摩尔比50。液体产物丙酸甲酯收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用甲苯作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0028]
实施例2
[0029]
在实施例2中,除了称取0.0212克三氯化铑替代0.0114克醋酸钯(ⅱ),其余的催化剂制备过程和反应过程皆与实施例1相同。金属负载量0.7wt%。具体反应结果参考表1。
[0030]
实施例3
[0031]
在实施例3中,除了称取0.0175克醋酸钴替代0.0114克醋酸钯(ⅱ),其余的催化剂制备过程和反应过程与实施例1相同。金属负载量0.5wt%。具体反应结果参考表1。
[0032]
实施例4
[0033]
在实施例4中,除了称取0.0366克乙酰丙酮铜(ⅱ)替代0.0114克醋酸钯(ⅱ),其余的催化剂制备过程与反应过程与实施例1相同。金属负载量0.9wt%。具体反应结果参考表1。
[0034]
实施例5
[0035]
在实施例5中,聚合物合成过程除了用二氯甲烷溶剂替代四氢呋喃溶剂,其余的催化剂制备过程与实施例1相同。
[0036]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入丙烯和co混合气(丙烯:co=1:2,体积比,下同),甲醇原料溶液经高压计量泵泵入反应器中开始反应,丙烯和甲醇制丁酸甲酯反应温度110℃,反应压力2.5mpa,甲醇液时空速0.12h-1
,co/甲醇摩尔比40。液体产物丁酸甲酯收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的
hp-7890n气相色谱分析,采用甲苯作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0037]
实施例6
[0038]
在实施例6中,聚合物合成过程除了搅拌12小时替代搅拌0.5小时,其余的催化剂制备过程与实施例1相同。
[0039]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入1-丁烯和co混合气(1-丁烯:co=1:6),甲醇原料经高压计量泵泵入反应器中开始反应,1-丁烯和甲醇制戊酸甲酯反应温度100℃,反应压力2.5mpa,甲醇液时空速0.12h-1
,co/甲醇摩尔比40。液体产物收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用甲醇作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0040]
实施例7
[0041]
在实施例7中,除了将自由基引发剂用过氧化二苯甲酰替代偶氮二异丁腈,其余的催化剂制备过程与实施例1相同。
[0042]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入1-丁烯和co混合气(1-丁烯:co=1:6),甲醇原料经高压计量泵泵入反应器中开始反应,1-丁烯和甲醇制戊酸甲酯反应温度105℃,反应压力2.5mpa,甲醇液时空速0.12h-1
,co/甲醇摩尔比40。液体产物收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用甲醇作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0043]
实施例8
[0044]
在实施例8中,除了将自由基引发剂偶氮二异丁腈0.1克替代为偶氮二异丁腈0.25克,其余的催化剂制备过程与实施例1相同。
[0045]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入co纯气,1-戊烯和甲醇溶液原料分别经高压计量泵泵入反应器中开始反应,1-戊烯和甲醇制己酸甲酯反应温度100℃,反应压力2mpa,1-戊烯和甲醇液时空速皆为0.1h-1
,co/甲醇摩尔比40。液体产物收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用甲苯作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0046]
实施例9
[0047]
在实施例8中,除了将自由基引发剂偶氮二异丁腈0.1克替代为偶氮二异丁腈0.5克,其余的催化剂制备过程与实施例1相同。
[0048]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入co纯气,1-戊烯和甲醇溶液原料分别经高压计量泵泵入反应器中开始反应,1-戊烯和甲醇制己酸甲酯反应温度105℃,反应压力2mpa,1-戊烯和甲醇液时空速皆为0.12h-1
,co/甲醇摩尔比40。液体产物收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用甲苯作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0049]
实施例10
[0050]
在实施例1中,除了将2.0g对乙烯基苯磺酸替代为2.0g乙烯基磺酸2g,其余的催化剂制备过程与实施例1相同。
[0051]
将上述制备的固体多相催化剂加入到滴流床反应器中,通入co纯气,1-己烯和甲醇溶液原料分别经高压计量泵泵入反应器中开始反应,1-己烯和甲醇制庚酸甲酯反应温度100℃,反应压力2mpa,1-己烯和甲醇液时空速皆为0.1h-1
,co/甲醇摩尔比40。液体产物收集于冷阱收集罐内。液体产物使用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用甲苯作内标。反应尾气使用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。具体反应结果参考表1。
[0052]
以上已对本发明进行了详细描述,但本发明并不局限于本文所描述具体实施方式。本领域技术人员理解,在不背离本发明范围的情况下,可以作出其他更改和变形。本发明的范围由所附权利要求限定。
[0053]
表1.烷氧基羰基化反应结果
[0054]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。