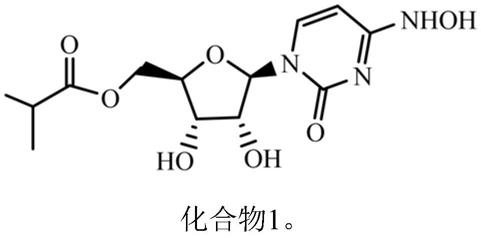
一种核糖核苷类似物的多晶型物、其制备方法及应用
1.本技术要求申请人于2021年04月23日向中国国家知识产权局提交的专利申请号202110440883.8发明名称为“一种核糖核苷类似物的多晶型物、其制备方法及应用”的在先申请的优先权。所述在先申请的全文通过引用的方式结合于本技术中。
技术领域
2.本发明属于化合物晶型领域,具体涉及一种核糖核苷类似物的多晶型物、其制备方法及应用。
背景技术:
3.molnupiravir(eidd-2801,cas号:2349386-89-4)是在美国亚特兰大(乔治亚州)的埃默里大学药物创新公司开发的药物。它是核糖核苷类似物,模仿天然存在的核苷,在单链rna病毒复制过程中产生错误来阻止病毒繁殖,从而抑制病毒的复制,其结构如下:
[0004][0005]
已有研究显示,eidd-2801对鼠类肝炎病毒(mhv)、严重急性呼吸综合征冠状病毒(sars-cov)、中东呼吸综合征冠状病毒(mers-cov)、新型冠状病毒(2019-ncov)等具有治疗效果。
[0006]
如何开发eidd-2801更适于成药的药物晶型,特别是使稳定性、吸湿性、保存性和/或药效等得到改善的晶型,从而在制药及用药阶段取得良好效果,成为亟待解决的技术问题。
技术实现要素:
[0007]
为了改善上述技术问题,本发明提供一种化合物1(eidd-2801)的多晶型物:
[0008][0009]
本发明提供化合物1的晶型i,所述晶型i使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在6.45
±
0.20
°
、12.99
±
0.20
°
、16.30
±
0.20
°
、16.95
±
0.20
°
、17.19
±
0.20
°
、21.21
±
0.20
°
处具有特征峰。
[0010]
优选地,所述晶型i使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在6.45
±
0.20
°
、12.99
±
0.20
°
、16.30
±
0.20
°
、16.95
±
0.20
°
、17.19
±
0.20
°
、21.21
±
0.20
°
、27.96
±
0.20
°
、28.39
±
0.20
°
处具有特征峰。
[0011]
还优选地,所述晶型i使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在6.45
±
0.20
°
、12.99
±
0.20
°
、16.30
±
0.20
°
、16.95
±
0.20
°
、17.19
±
0.20
°
、19.60
±
0.20
°
、20.53
±
0.20
°
、21.21
±
0.20
°
、27.96
±
0.20
°
、28.39
±
0.20
°
、33.94
±
0.20
°
处具有特征峰。
[0012]
更优选地,所述晶型i使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在6.48
±
0.20
°
、12.99
±
0.20
°
、16.30
±
0.20
°
、16.95
±
0.20
°
、17.19
±
0.20
°
、18.03
±
0.20
°
、19.35
±
0.20
°
、19.60
±
0.20
°
、20.31
±
0.20
°
、20.53
±
0.20
°
、21.21
±
0.20
°
、27.96
±
0.20
°
、28.39
±
0.20
°
、28.92
±
0.20
°
、31.58
±
0.20
°
、33.94
±
0.20
°
处具有特征峰。
[0013]
根据本发明的实施方案,所述晶型i使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射如表1所示,误差范围
±
0.20
°
:
[0014]
表1晶型i的xrpd解析数据
[0015]
峰编号2θ[
°
]相对强度%峰编号2θ[]相对强度%16.451100.02528.88012.929.73016.02629.6596.0312.99055.82730.08115.6416.30057.42830.8685.4516.95054.22931.63914.7617.18949.93033.0526.6718.01027.83133.42012.3818.2905.73233.94120.9919.38019.63334.3734.31019.60022.83436.1986.61119.84012.83536.5194.11220.26014.33637.2712.71320.53028.43737.8394.51421.21064.03839.1187.71522.5295.73940.1684.21622.9195.44040.4205.21723.31916.34140.93017.81823.91012.24242.0726.61924.19911.94342.3294.92025.6999.64443.5024.92126.34913.64544.11015.62227.4694.64645.1904.62327.96140.74747.4263.42428.38943.7
ꢀꢀꢀ
。
[0016]
优选地,所述晶型i具有基本如图1所示的粉末x射线衍射图。
[0017]
根据本发明的实施方案,所述晶型i具有基本如图2所示的dsc谱图。
[0018]
根据本发明的实施方案,所述晶型i具有基本如图3所示的tga谱图。
[0019]
本发明提供化合物1的晶型ii,所述晶型ii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在3.20
±
0.20
°
、16.94
±
0.20
°
、17.98
±
0.20
°
、20.50
±
0.20
°
、21.24
±
0.20
°
、27.94
±
0.20
°
处具有特征峰。
[0020]
优选地,所述晶型ii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在3.20
±
0.20
°
、6.44
±
0.20
°
、16.94
±
0.20
°
、17.98
±
0.20
°
、20.50
±
0.20
°
、20.28
±
0.20
°
、21.24
±
0.20
°
、27.94
±
0.20
°
处具有特征峰。
[0021]
更优选地,所述晶型ii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在3.20
±
0.20
°
、6.44
±
0.20
°
、16.94
±
0.20
°
、17.98
±
0.20
°
、19.36
±
0.20
°
、20.50
±
0.20
°
、20.28
±
0.20
°
、21.24
±
0.20
°
、27.94
±
0.20
°
、28.36
±
0.20
°
处具有特征峰。
[0022]
根据本发明的实施方案,所述晶型ii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射如表2所示,误差范围
±
0.20
°
:
[0023]
表2晶型ii的xrpd解析数据
[0024]
峰编号2θ[
°
]相对强度%峰编号2θ[
°
]相对强度%13.198100.02733.9008.526.44017.02834.3626.739.7002.72934.9561.8412.9819.93035.1751.6516.2978.83136.1383.1616.94080.33236.5793.2717.98032.63337.2962.9818.2995.83437.8181.4919.36016.23538.6842.21019.8199.33639.0962.21120.27818.53740.4192.21220.50134.43840.8633.61321.24168.13942.3372.61422.5195.04043.5391.61523.3178.14144.1772.81624.1417.14245.2621.01725.6963.34348.3971.31826.3594.4
ꢀꢀꢀ
1927.4374.2
ꢀꢀꢀ
2027.94251.4
ꢀꢀꢀ
2128.36014.9
ꢀꢀꢀ
2228.83910.6
ꢀꢀꢀ
2330.06112.1
ꢀꢀꢀ
2430.8222.3
ꢀꢀꢀ
2531.54211.1
ꢀꢀꢀ
2633.6626.3
ꢀꢀꢀ
。优选地,所述晶型ii具有基本如图4所示的粉末x射线衍射图。
[0025]
根据本发明的实施方案,所述晶型ii具有基本如图5所示的dsc谱图。
[0026]
根据本发明的实施方案,所述晶型ii具有基本如图6所示的tga谱图。
[0027]
本发明提供化合物1的晶型iii,所述晶型iii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在16.71
±
0.20
°
、17.57
±
0.20
°
、18.22
±
0.20
°
、19.90
±
0.20
°
、22.31
±
0.20
°
处具有特征峰。
[0028]
优选地,所述晶型iii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在14.15
±
0.20
°
、16.71
±
0.20
°
、17.57
±
0.20
°
、18.22
±
0.20
°
、19.90
±
0.20
°
、20.52
±
0.20
°
、22.31
±
0.20
°
、24.39
±
0.20
°
处具有特征峰。
[0029]
更优选地,所述晶型iii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射在10.10
±
0.20
°
11.10
±
0.20
°
、14.15
±
0.20
°
、16.71
±
0.20
°
、17.57
±
0.20
°
、18.22
±
0.20
°
、19.90
±
0.20
°
、20.52
±
0.20
°
、22.31
±
0.20
°
、24.13
±
0.20
°
、24.39
±
0.20
°
、25.42
±
0.20
°
处具有特征峰。
[0030]
根据本发明的实施方案,所述晶型iii使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射如表3所示,误差范围
±
0.20
°
:
[0031]
表3晶型iii的xrpd解析数据
[0032]
峰编号2θ[
°
]相对强度%峰编号2θ[
°
]相对强度%110.10020.31928.89016.6211.10119.62030.0694.4313.21113.62130.9409.3414.15027.92231.2394.7516.71049.82331.8705.9617.57088.72432.39016.3718.220100.02533.8706.8819.90041.82635.1294.2920.28017.52736.6983.51020.52033.32837.1395.01120.87017.02938.6304.41222.31042.53039.0604.51322.7108.03140.9703.91423.3995.13241.6107.61524.13020.43342.5919.01624.39029.83445.6902.51725.42121.03546.6822.91827.16113.83647.4792.9
[0033]
优选地,所述晶型iii具有基本如图7所示的粉末x射线衍射图。
[0034]
根据本发明的实施方案,所述晶型iii具有基本如图8所示的dsc谱图。
[0035]
根据本发明的实施方案,所述晶型iii具有基本如图9所示的tga谱图。
[0036]
根据本发明的实施方案,所述多晶型物包括化合物1的非溶剂合物(无水合物)以及溶剂合物的晶型形式;在一些实施方案中,所述多晶型包括水合物,例如为半水合物、一水合物、二水合物、三水合物、四水合物。
[0037]
本发明还提供上述化合物1(eidd-2801)的多晶型物的制备方法,包括将所述化合物1和溶剂混合,经重结晶,制备得到所述多晶型物;
[0038]
所述溶剂选自有机溶剂和/或水。
[0039]
根据本发明的实施方案,所述有机溶剂选自甲醇、乙醇、异丙醇、丙酮、乙腈、甲苯、乙酸乙酯、乙酸丁酯、甲基叔丁基醚、四氢呋喃等中的一种、两种或更多种。
[0040]
根据本发明的实施方案,所述溶剂可以选自甲醇、乙醇、异丙醇、丙酮、乙腈、甲苯、乙酸乙酯、乙酸丁酯、四氢呋喃、乙酸乙酯和甲基叔丁基醚的混合溶剂、乙醇和水的混合溶剂、或者乙腈和水的混合溶剂。
[0041]
根据本发明的实施方案,所述化合物1与溶剂的质量体积比为1g:(2-10)ml,例如为5g:25ml。
[0042]
根据本发明的实施方案,所述制备方法包括:将所述化合物1和溶剂混合,升温搅拌至体系溶清,降温、洗涤、干燥,得到所述多晶型物。
[0043]
根据本发明的实施方案,所述多晶型物的制备方法可以选自下述任一种方法:
[0044]
方法一:化合物1和乙酸乙酯按照质量体积比为1g:(2-10)ml混合,加热搅拌升温至回流(优选回流时间1-5小时),缓慢降温至10-15℃,过滤,用乙酸乙酯淋洗,干燥(优选干燥温度30-50℃),得到所述多晶型物;
[0045]
方法二:化合物1和乙酸乙酯按照质量体积比为1g:(2-10)ml混合,加热搅拌升温至回流(优选回流0.5-2小时),降温至50-55℃,向体系中加入(优选滴加)甲基叔丁基醚(优选2min滴完);继续降温至10-15℃,搅拌(优选搅拌0.5-1小时),过滤,洗涤,干燥,得到所述多晶型物;优选地,乙酸乙酯和甲基叔丁基醚的体积比为1:(0.8-1.3);
[0046]
方法三:化合物1、无水乙醇与水按照质量体积比1g:(2-10)ml:(0.3-0.8)ml,加热搅拌升温至体系溶清,缓慢降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0047]
方法四:化合物1和无水乙醇按照质量体积比为1g:(2-10)ml混合,加热搅拌升温至体系溶清,冰水浴降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0048]
方法五:化合物1和丙酮按照质量体积比为1g:(2-10)ml混合,升温回流打浆,然后缓慢降温至0~5℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0049]
方法六:化合物1和甲醇按照质量体积比为1g:(2-10)ml混合,升温搅拌至体系溶清,然后缓慢降温至0~5℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0050]
方法七:化合物1和水按照质量体积比为1g:(2-10)ml混合,加热搅拌升温至体系溶清(例如加热至60℃),自然降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0051]
方法八:化合物1和四氢呋喃按照质量体积比为1g:(2-10)ml混合,加热搅拌升温至体系溶清(例如升温至60℃),自然降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0052]
方法九:化合物1、水和乙腈按照质量体积比为1g:(0.3-0.8)ml:(0.8-2)ml升温搅拌至体系溶清,然后缓慢降温至0~5℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0053]
方法十:化合物1、水和甲醇按照质量体积为1g:(0.3-0.8)ml:(0.8-2)ml升温搅拌至体系溶清,然后缓慢降温至0~5℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0054]
方法十一:化合物1和四氢呋喃按照质量体积比为1g:(2-10)ml混合,加热搅拌升温至体系溶清,自然降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0055]
方法十二:化合物1和甲苯按照质量体积比为1g:(2-10)ml混合,加热搅拌回流,自然降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0056]
方法十三:化合物1和异丙醇按照质量体积比为1g:(2-10)ml混合,加热搅拌至体系溶清(例如加热至70℃),自然降温至10-15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0057]
方法十四:化合物1和异丙醇按照质量体积比为1g:(2-10)ml混合,加热搅拌至体系溶清,然后缓慢降温至10~15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物;
[0058]
方法十五:化合物1和正丙醇按照质量体积比为1g:(2-10)ml混合,加热搅拌至体系溶清,然后缓慢降温至10~15℃,搅拌,过滤,洗涤,干燥,得到所述多晶型物。
[0059]
本发明还提供一种药物组合物,其含有上述多晶型物。
[0060]
根据本发明的实施方案,所述药物组合物还可以包括至少一种药学上可接受的辅料。
[0061]
根据本发明的实施方案,所述药物组合物可以为本领域已知制剂。例如为口服剂、胶囊剂、锭剂、注射剂等。
[0062]
本发明还提供上述多晶型物或药物组合物在制备治疗抗病毒药物中的应用。例如,所述抗病毒药物可以选自冠状病毒和/或鼠类肝炎病毒(mhv),例如所述冠状病毒可以选自严重急性呼吸综合征冠状病毒(sars-cov)、中东呼吸综合征冠状病毒(mers-cov)和/或新型冠状病毒(2019-ncov)。
[0063]
有益效果
[0064]
本发明提供的晶型纯度高,具有良好的稳定性和耐保存性,能够在加速实验、高温(例如50
±
2℃、60
±
2℃、70
±
2℃)、高湿(例如湿度70
±
5%rh,80
±
5%rh,90
±
5%rh)、光照(例如总光照度不低于1.0
×
106lux
·
hr)条件下,长期保存,不易发生转晶,且杂质(单杂和/或总杂)含量变化小、甚至基本无明显变化。有利于进一步应用于在制剂过程中提高稳定性和安全性,降低副作用,增加生物利用度等。
[0065]
本发明晶型的制备方法成本低廉,操作简单,反应条件容易控制,可以稳定地获得目标晶型。
附图说明
[0066]
图1为晶型i的粉末x射线衍射图谱;
[0067]
图2为晶型i的dsc谱图;
[0068]
图3为晶型i的tga谱图;
[0069]
图4为晶型ii的粉末x射线衍射图;
[0070]
图5为晶型ii的dsc谱图;
[0071]
图6为晶型ii的tga谱图;
[0072]
图7为晶型iii的粉末x射线衍射图;
[0073]
图8为晶型iii的dsc谱图;
[0074]
图9为晶型iii的tga谱图。
具体实施方式
[0075]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0076]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0077]
粉末x射线衍射的扫描范围4.0-50.0
°
,0.01/1s,cu(40kv,100ma)。
[0078]
实施例1
[0079]
称取eidd-2081粗品5g于反应瓶中,加25ml乙酸乙酯,加热搅拌升温至回流,回流3小时,缓慢降至10-15℃,过滤,并用少量乙酸乙酯淋洗。刮出,置于45℃鼓风烘箱中干燥24h,得到产品。产品收率95%。
[0080]
对实施例产品进行鉴定,得到晶型i。
[0081]
实施例2
[0082]
称取eidd-2081粗品5g于反应瓶中,加25ml无水乙醇与2.5ml水,加热搅拌升温至70℃,固体溶清,缓慢冷却至10-15℃,搅拌0.5h,过滤,并用少量乙醇淋洗。刮出,置于50℃鼓风烘箱中干燥24h,得到产品。产品收率75%。
[0083]
对实施例产品进行鉴定,得到晶型i。
[0084]
实施例3
[0085]
称取eidd-2081粗品5g于反应瓶中,加25ml无水乙醇,加热搅拌升温至70℃,固体溶清,冰水浴降温降至10-15℃,搅拌0.5h,过滤,并用少量冰乙醇淋洗。刮出,置于50℃鼓风烘箱中干燥24h,得到产品。产品收率78%。
[0086]
对实施例产品进行鉴定,得到晶型i。
[0087]
实施例4
[0088]
称取eidd-2081粗品5g加到反应瓶中,并加入30ml丙酮升温回流打浆2h,然后缓慢降温至0~5℃搅拌1h,过滤,少量丙酮洗涤滤饼,进干燥箱干燥(t=50℃)得到白色固体3.52g。产品收率为70.4%。
[0089]
对实施例产品进行鉴定,得到晶型i。
[0090]
实施例5
[0091]
称取eidd-2081粗品5g加到反应瓶中,并加入2.5ml水和5ml乙腈升温溶清搅拌2h,然后缓慢降温至0~5℃搅拌1h,过滤,少量乙腈洗涤滤饼,进干燥箱干燥(t=50℃)得到白色固体2.23g。产品收率为44.6%。
[0092]
对实施例产品进行鉴定,得到晶型i。
[0093]
实施例6
[0094]
称取eidd-2081粗品5g于反应瓶中,加20ml四氢呋喃,加热搅拌升温至60℃,固体
溶清,自然降温至10-15℃,搅拌1h,过滤,少量冰四氢呋喃淋洗,刮出,置于50℃鼓风烘箱中干燥24h,得到晶型ii。收率60%。
[0095]
实施例7
[0096]
称取eidd-2081粗品5g加到反应瓶中,并加入25ml乙醇升温溶清搅拌2h,然后在保温条件下缓慢降温,温度降至60℃时,有固体析出,在该温度下保温0.5小时,并继续缓慢降温。每半小时降温5℃,降温至35℃时用冰水冷却,并继续降温至10~15℃搅拌1h,过滤,少量乙醇洗涤滤饼,进干燥箱干燥(t=50℃)。
[0097]
实施例8
[0098]
还能够通过下述任一种制备方法得到eidd-2081的多晶型物。
[0099]
称取eidd-2081粗品5g于反应瓶中,加20ml乙酸乙酯,加热搅拌升温至回流,回流0.5小时,降温至50-55℃,滴加20ml甲基叔丁基醚,2min滴完。继续降温至10-15℃,搅拌0.5h,过滤,并用少量冰甲基叔丁基醚淋洗。刮出,置于45℃鼓风烘箱中干燥24h,产品收率95%。
[0100]
或者,称取eidd-2081粗品5g加到反应瓶中,并加入10ml甲醇升温溶清搅拌2h,然后缓慢降温至0~5℃搅拌1h,过滤,少量甲醇洗涤滤饼,进干燥箱干燥(t=50℃)得到白色固体2.53g,收率=50.6%。
[0101]
或者,称取eidd-2081粗品5g于反应瓶中,加10ml水,加热搅拌升温至60℃,固体溶清,自然降温至10-15℃,搅拌1h,过滤,刮出,置于50℃鼓风烘箱中干燥24h。收率76%。
[0102]
或者,称取eidd-2081粗品5g加到反应瓶中,并加入2.5ml水和5ml甲醇升温溶清搅拌2h,然后缓慢降温至0~5℃搅拌1h,过滤,少量甲醇洗涤滤饼,进干燥箱干燥(t=50℃)得到白色固体2.17g,收率=43.4%。
[0103]
或者,称取eidd-2081粗品5g于反应瓶中,加50ml乙酸丁酯,加热搅拌升温至回流1h,自然降温至10-15℃,搅拌1h,过滤,少量冰乙酸丁酯淋洗,刮出,置于50℃鼓风烘箱中干燥24h。收率65%。
[0104]
或者,称取eidd-2081粗品5g于反应瓶中,加50ml甲苯,加热搅拌升温至回流1h,自然降温至10-15℃,搅拌1h,过滤,少量冰甲苯淋洗,刮出,置于50℃鼓风烘箱中干燥24h。收率70%。
[0105]
或者,称取eidd-2081粗品5g于反应瓶中,加50ml异丙醇,加热搅拌升温至70℃,固体溶清,自然降温至10-15℃,搅拌1h,过滤,少量冰异丙醇淋洗,刮出,置于50℃鼓风烘箱中干燥24h,收率71%。
[0106]
或者,称取eidd-2081粗品5g于反应瓶中,加50ml正丙醇,加热搅拌升温至70℃,固体溶清,自然降温至10-15℃,搅拌1h,过滤,少量冰正丙醇淋洗,刮出,置于50℃鼓风烘箱中干燥24h,收率65%。
[0107]
实施例9晶型鉴定试验
[0108]
晶型i的xrd谱图如图1所示,谱图解析数据如表1所示。晶型i的dsc谱图和tga谱图分别如图2和图3所示。
[0109]
晶型ii的xrd谱图如图4所示,谱图解析数据如表2所示。晶型ii的dsc谱图和tga谱图分别如图5和图6所示。
[0110]
晶型iii的xrd谱图如图7所示,谱图解析数据如表3所示。晶型iii的dsc谱图和tga
谱图分别如图8和9所示。
[0111]
表1晶型i的xrpd解析数据
[0112]
峰编号2θ[
°
]相对强度%峰编号2θ[
°
]相对强度%16.451100.02528.88012.929.73016.02629.6596.0312.99055.82730.08115.6416.30057.42830.8685.4516.95054.22931.63914.7617.18949.93033.0526.6718.01027.83133.42012.3818.2905.73233.94120.9919.38019.63334.3734.31019.60022.83436.1986.61119.84012.83536.5194.11220.26014.33637.2712.71320.53028.43737.8394.51421.21064.03839.1187.71522.5295.73940.1684.21622.9195.44040.4205.21723.31916.34140.93017.81823.91012.24242.0726.61924.19911.94342.3294.92025.6999.64443.5024.92126.34913.64544.11015.62227.4694.64645.1904.62327.96140.74747.4263.42428.38943.7
ꢀꢀꢀ
[0113]
表2晶型ii的xrpd解析数据
[0114][0115][0116]
表3晶型iii的xrpd解析数据
[0117]
峰编号2θ[
°
]相对强度%峰编号2θ[
°
]相对强度%110.10020.31928.89016.6211.10119.62030.0694.4313.21113.62130.9409.3414.15027.92231.2394.7516.71049.82331.8705.9617.57088.72432.39016.3718.220100.02533.8706.8819.90041.82635.1294.2920.28017.52736.6983.51020.52033.32837.1395.01120.87017.02938.6304.4
1222.31042.53039.0604.51322.7108.03140.9703.91423.3995.13241.6107.61524.13020.43342.5919.01624.39029.83445.6902.51725.42121.03546.6822.91827.16113.83647.4792.9
[0118]
实施例10晶型性能测试
[0119]
(一)溶解度
[0120]
考察试剂:纯化水、甲醇、dmf、乙酸乙酯、丙酮、二氯甲烷、乙腈、乙醇;
[0121]
实验步骤:
[0122]
室温下,取研磨成细粉的供试品(即晶型i、晶型ii和晶型iii),按照不同溶解度要求称取定量供试品(见表1-表3)于25℃水浴中震摇,每隔5分钟强力震摇30秒,观察30分钟内的溶解情况,无目视可见的溶质颗粒,即视为完全溶解。
[0123]
表1
[0124][0125]
表2
[0126][0127][0128]
表3
[0129][0130]
晶型i-iii在不同溶剂中的溶解度汇总如表4所示。
[0131]
表4
[0132][0133]
(二)引湿性
[0134]
实验仪器:具塞玻璃称量瓶(外径50mm,高为15mm)。
[0135]
实验步骤:
[0136]
1、取干净的具塞玻璃称量瓶于实验前一天置于适宜的25℃恒温干燥器(下部放置氯化铵或硫酸铵饱和溶液)精密称定其重量(m1);
[0137]
2、取供试品适量,平铺于上述称量瓶中,供试品厚度一般约为1mm,精密称定重量(m2);
[0138]
3、将称量瓶敞口,并与瓶盖同置于上述恒温恒湿条件下24小时;
[0139]
4、盖好称量瓶盖子,精密称定重量(m3)。
[0140][0141]
结果如表5所示,晶型i-iii略有引湿性。
[0142]
表5
[0143][0144][0145]
(三)稳定性
[0146]
光照稳定性:将晶型i-iii分别置于光照强度4500lx条件下,于第5天和第12天取样,用高效液相色谱(hplc)检测纯度。
[0147]
高湿稳定性:将晶型i-iii分别置于室温、相对湿度92.5%
±
5%的条件下,于第11天和第30天取样,用hplc检测纯度。
[0148]
热稳定性:将晶型i-iii分别置于温度60℃
±
2℃的条件下,于第11天和第30天取样,用hplc检测纯度。
[0149]
加速稳定性:将晶型i-iii分别置于温度40℃
±
2℃、湿度75%
±
5%的条件下,于第1个月和第2个月取样,用hplc检测纯度,用xrpd检测晶型。
[0150]
其中,hplc检测的条件如下:
[0151]
1.1色谱条件
[0152]
色谱柱:bds hypersilc18,250
×
4.6mm,5μm
[0153]
柱温:40℃
[0154]
流速:1.0ml/min
[0155]
检测器波长:254nm
[0156]
进样量:10μl
[0157]
流动相a:0.01mol/l的磷酸二氢钾溶液
[0158]
流动相b:甲醇
[0159]
梯度洗脱:
[0160]
[0161][0162]
1.2溶液配制
[0163]
空白溶液:5%甲醇
[0164]
杂质对照品储备溶液:分别精密称取10mg杂质(杂质imp-g,杂质尿苷,杂质imp-d,杂质imp-b,)至10ml容量瓶中,加5%甲醇超声溶解并稀释至刻度,摇匀各杂质储备液(1mg/ml)。
[0165]
供试品溶液:精密称取25mg供试品(即晶型i-iii),置25ml容量瓶中,加5%甲醇超声溶解并稀释至刻度,摇匀,即得(1mg/ml)。
[0166]
对照品溶液:精密称取25mg对照品,置25ml容量瓶中,加5%甲醇超声溶解并稀释至刻度,摇匀,即得对照品储备液1;精密移取1ml对照品储备液1置100ml容量瓶中,加5%甲醇稀释至刻度,摇匀,即得对照品储备液2;精密移取1ml对照品储备液2置10ml容量瓶中,加5%甲醇稀释至刻度,摇匀,即得对照品溶液(1μg/ml)。
[0167]
系统适应性溶液:精密称取eidd-2801对照品50mg,至50ml容量瓶中,向其中分别加入0.5ml的各杂质储备液,加5%甲醇超声溶解稀释至刻度,摇匀,即得(eidd-2801对照品1mg/ml,各杂质10μg/ml)。
[0168]
稳定性测试结果如表6-8所示。
[0169]
表6
[0170][0171][0172]
表7
[0173][0174]
表8
[0175][0176][0177]
从稳定性的试验结果来看,晶型i-iii均具有良好的稳定性。加速试验两个月后对测试样品的晶型进行检测(经xrpd鉴定),晶型未发生变化。
[0178]
由此可见,本发明晶型的纯度高,杂质含量低;具有良好的稳定性和耐保存性,能够在加速实验、高湿、高温、光照条件下,长期保存,不易发生转晶,杂质含量变化小、甚至基本无明显变化。
[0179]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡
在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。