1.本发明涉及生物工程领域,涉及一种提高线性多肽生物表达的方法,具体涉及一种新型的融合表达标签,以及在促进异源多肽表达中的应用。
背景技术:
2.大肠杆菌表达系统具有较高的表达水平,较快的生长速度,适合连续和高细胞密度的培养方法,因此大肠杆菌是迄今为止生物技术中最受欢迎的重组蛋白生产宿主。然而,大肠杆菌表达系统仍存在一些不足,其中一个限制就是小蛋白和多肽的生产。由于通过直接表达方式来表达较小片段的多肽时,容易出现降解,导致产量低,同时也不适合表达对菌体生长不利的毒肽或抗菌肽。另外,许多重组多肽在表达过程中由于翻译速度过快同时缺少同源伴侣而容易在细菌中产生错误折叠。为了解决这一问题,许多标签被融合到目标蛋白中,其中一些通过形成包涵体蛋白,增加蛋白的产量。与可溶性表达相比,包涵体表达具有产量高、稳定性强、易于分离纯化等优势,而且减轻了目的蛋白或多肽对宿主细胞的毒性作用,但是包涵体蛋白需要通过复杂的复性过程才能得到正确折叠的功能蛋白,这在一定程度抵消了包涵体高表达优势。另外,对于生物医学应用来说,蛋白标签的去除是非常关键的。
技术实现要素:
3.发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种多肽表达的方法。
4.本发明还要解决的技术问题是提供一种制备多肽的方法。
5.本发明还要解决的技术问题是提供一种eddie(c69e、c168e)-sumo联合标签。
6.本发明还要解决的技术问题是提供编码上述联合标签的基因。
7.本发明还要解决的技术问题是提供一种蛋白表达载体。
8.本发明还要解决的技术问题是提供一种试剂盒。
9.为了解决上述第一个技术问题,本发明公开了一种多肽表达的方法,包括以下步骤:将eddie(c69e、c168e)-sumo联合标签与目标多肽融合表达所述目标多肽。
10.其中,所述eddie(c69e、c168e)-sumo联合标签包括seq id no.1所示的氨基酸序列。
11.其中,本发明所述的eddie标签为包涵体标签蛋白标签;所述的eddie标签是npro猪瘟自剪切蛋白的npro突变蛋白,分别将其69位的cys突变为glu,168位的cys突变为glu。这主要是由于天然npro蛋白纯化蛋白存在许多不足,与之相比,本发明所提供的npro突变蛋白eddie可溶性增加,具有更快复性和更高剪切效率,但eddie在自剪切效率方面仍存在一定的不足;而在将eddie的剪切活性关键位点69位和168位突变后,使其不具有自剪切能力,但保留形成包涵体能力。
12.其中,所述目标多肽为抗菌肽、线性肽和毒肽中的任意一种。
13.为了解决上述第二个技术问题,本发明公开了一种制备多肽的方法,采用eddie(c69e、c168e)和sumo这两种蛋白标签建立c端融合表达体系,同时用sumo蛋白酶特异性的切割来去除标签,从而得到目的多肽,最后酶切产物通过固相萃取方法纯化。其中,本发明通过所建立的c端融合表达体系,将eddie的剪切活性关键位点69位和168位突变,使其不具有自剪切能力,但保留形成包涵体能力;利用sumo蛋白标签还具有的促进蛋白的正确折叠的能力来使eddie复性过程中不容易沉淀。
14.具体地,所述方法包括以下步骤:将eddie(c69e、c168e)-sumo联合标签与目标多肽融合表达,得到融合蛋白;将所述融合蛋白复性;采用与所述联合标签相适应的特异蛋白酶切除复性后融合蛋白中的sumo标签,纯化,即得所述目标多肽。
15.其中,所述eddie(c69e、c168e)-sumo联合标签包括seq id no.1所示的氨基酸序列。
16.其中,所述目标多肽为线性肽,包括但不限于抗菌肽、线性肽和毒肽中的任意一种。
17.其中,所述复性过程中采用的缓冲液为1-1.5m tris-hcl,ph 7-8,2.5-10%甘油,1-10mm三(2-羧乙基)膦盐酸盐(tcep盐酸盐),优选为1m tris-hcl,ph 7.8,5%甘油,2mm tcep盐酸盐。
18.其中,所述复性为透析复性。
19.其中,所述联合标签相适应的特异蛋白酶为sumo蛋白酶,其效率较高。
20.其中,所述切除复性后融合蛋白中的sumo标签方法为的剪切方法为:直接向浓度为0.8-1.2mg复性融合蛋白/ml复性液复性融合蛋白中添加相应量的sumo蛋白酶;所述sumo蛋白酶的用量为待剪切的蛋白量
×
0.007
÷
sumo蛋白酶浓度。
21.其中,所述酶切为在25-35℃反应2.5-4.5h,优选为30℃反应3.5h。
22.其中,所述纯化为采用c8反相柱纯化。
23.其中,所述c8反相柱的参数为碳含量:9%,比表面积:280m2/g粒径:40-75μm平均孔径:
24.为了解决上述第三个技术问题,本发明公开了一种eddie(c69e、c168e)-sumo联合标签,其包括seq id no.1所示的氨基酸序列。
25.为了解决上述第四个技术问题,本发明公开了一种编码上述eddie(c69e、c168e)-sumo联合标签的基因,其包括seq id no.2所示的核苷酸序列。
26.为了解决上述第五个技术问题,本发明公开了一种蛋白表达载体,其包括上述eddie(c69e、c168e)-sumo联合标签。
27.为了解决上述第五个技术问题,本发明公开了一种试剂盒,其包括上述eddie(c69e、c168e)-sumo联合标签,或上述编码eddie(c69e、c168e)-sumo联合标签的基因,或上述蛋白表达载体。
28.有益效果:与现有技术相比,本发明具有如下优势:
29.本发明涉及一种线性多肽表达和纯化的方法,将多肽与包含体蛋白eddie(c69e、c168e)和sumo蛋白标签融合,这种融合多肽可以在大肠杆菌中以包含体形式表达纯化,并在透析复性后利用sumo蛋白酶将目的多肽切下,最后利用c8固相萃取反相柱纯化小肽,通过hplc色谱分析纯度在80%-90%;该方法可以有效解决包涵体难复性以及标签去除的问
题。
附图说明
30.下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
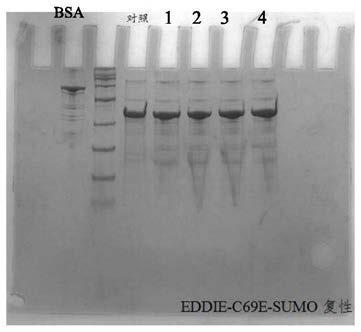
31.图1为eddie(c69e)-sumo复性剪切情况,对照为未复性的eddie(c69e)-sumo蛋白,1-4分别是复性的eddie(c69e)-sumo蛋白。
32.图2为eddie(c168e)-sumo复性剪切情况,对照为未复性的eddie(c168e)-sumo蛋白,1-4分别是复性的eddie(c168e)-sumo蛋白。
33.图3为载体构建rcr扩增电泳检测图。
34.图4为融合多肽酶切效果图。
35.图5为固相萃取小柱纯化效果图。
36.图6为多肽冻干成品图。
具体实施方式
37.下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
38.实施例1:
39.(1)构建氨基酸序列如seq id no.1所示的eddie(c69e、c168e)-sumo联合标签
40.以seq id no.3-24所示引物来基因合成seq id no.1所示的基因序列,序列预期长度分别约885bp。基因合成分为2轮进行:第一轮pcr反应体系(50μl):10μl phusion buffer(5
×
),1μl dntp mixture(10mmol/l),引物mix(seq id no.3和seq id no.24)2μl,0.5μl phusion酶(5u/μl),36.5μl h2o。第一轮pcr反应程序为98℃预变性30sec;18个循环:98℃,10sec;58℃,20sec;72℃,1.5min;72℃5min。第二轮pcr反应体系(50μl):10μl phusion buffer(5
×
),1μl dntp mixture(10mmol/l),eddie(c69ec168e)-sumo-1:1μl(10μmol/l),eddie(c69ec168e)-sumo-22:1μl(10μmol/l),第一轮模板1μl,0.5μl phusion酶(5u/μl),35.5μl h2o。第二轮pcr反应程序为98℃预变性30sec;25个循环:98℃,10sec;58℃,20sec;72℃,1.5min;72℃,5min。
41.pcr产物经1.5%琼脂糖凝胶电泳检测并通过挂柱pcr产物纯化试剂盒采用硅胶膜吸附dna的方法回收:1.将pcr反应液转移至一干净的1.5ml离心管中,加入3倍体积的buffer b3,充分混匀。2.将混合液全部移入吸附柱8000
×
g离心30sec。3.倒掉收集管中的液体,向吸附柱中加入500μl wash solution,9000
×
g离心30sec,倒掉收集管中的液体。4.重复步骤3一次。5.将空吸附柱和收集管放入离心机,9000
×
g离心1min。6.在吸附膜中央加入40μlelution buffer,室温静置l-2min,9000
×
g离心1min。elution buffer为2.5mm tris-hci,ph 8.5。然后将纯化后的pcr产物与ncoi/xhoi双酶切后的pet-15bvector进行连接,转化到大肠杆菌top10感受态细胞中,进行氨苄抗性筛选,挑取阳性转化子后提取质粒送本公司测序,测序结果显示序列正确,核苷酸序列如seq id no.25所示。
42.所述联合标签中第69位的cys突变为glu,第168位的cys突变为glu,图1和图2分别为eddie(c69e)-sumo和eddie(c168e)-sumo复性剪切情况,将eddie(c69e)-sumo和eddie
(c168e)-sumo蛋白透析于0.2m arg,20mm tris,0.01%tween-20,250mm nacl,5%甘油,5mm dtt,10mm硫酸镁,ph:9.0中,4℃过夜后分别取20μl进行蛋白电泳电泳,对照为透析未处理。证明69位和168位突变后不能发生自剪切反应,这两个突变在自剪切中发挥关键作用,本发明将两个位点均突变。
43.(2)将所述联合标签载体与目标多肽融合表达所述目标多肽,得到融合蛋白
44.扩增eddie(c69e、c168e)-sumo联合标签载体(图3,泳道1-2),得到线性化的标签载体片段:扩增引物为:f:5
′‑
taactcgaggatccggctgctaac-3
′
(seq id no.26),r:5
′‑
gcctccaatctgttcgcggtgagcctca-3
′
(seq id no.27),序列预期长度约为6497bp。pcr反应体系(50μl):10μl phusion buffer(5
×
),1μl dntp mixture(10mmol/l),上游引物1μl(10μmol/l),下游引物1μl(10μmol/l),pet15b-eddie(c69e、c168e)-sumo模板1μl,0.5μlphusion酶(5u/μl),35.5μl h2o。pcr反应程序为98℃预变性30sec;25个循环:98℃,10sec;58℃,20sec;72℃,1.5min;72℃,5min。
45.扩增第一目标多肽,如图3所示,泳道3为b147-1(28aa),其核苷酸序列分别为如seq id no.28所示,所述目标多肽的氨基酸序列如seq id no.29所示;扩增第二目标多肽,如图3所示,泳道4为glp1-1(31aa),其核苷酸序列分别为如seq id no.30所示,所述目标多肽的氨基酸序列如seq id no.31所示;扩增第三目标多肽,如图3所示,泳道5为cherry50(51aa),其核苷酸序列分别为如seq id no.32所示,所述目标多肽的氨基酸序列如seq id no.33所示;扩增第四目标多肽,如图3所示,泳道6为cherry80(80aa),其核苷酸序列分别为如seq id no.34所示,所述目标多肽的氨基酸序列如seq id no.35所示。
46.将目标多肽1~4的基因片段分别与线性化标签载体进行酶联(50℃20min),酶连体系为20μl体系:gibson重组酶:5μl,目标多肽基因片段:10ng,线性化标签载体:50ng,h2o:补齐到20μl。表达鉴定后送本公司测序,测序结果显示序列正确;成功构建重组融合多肽载体。
47.将所述融合多肽载体于大肠杆菌中诱导表达所述目标蛋白,1)活化:将所述融合多肽载体涂布于lb平板(amp ),37℃恒温培养箱过夜培养。第二天早上挑一个克隆至含2ml lb液(amp )的试管中,37℃,200rpm振摇至od600值约为0.6。2)将2ml菌液加入200ml lb中37℃,200rpm振摇至od600值约为0.6。3)诱导表达:加入iptg至终浓度为1mm,继续摇3~4h。4)细胞收集:离心(5000rpm,5min,4℃)。5)重悬洗涤:用washing buffer:50mm tris-hcl,5mm edta,1%triton x-100,ph 8.0悬浮细菌。6)超声破碎细胞。7)收集包涵体沉淀:离心(12,000rpm,15min,4℃),去上清。8)包涵体洗涤:washing buffer:50mm tris-hcl,5mm edta,1%triton x-100,ph 8.0重悬洗涤。9)包涵体溶解:用denaturation buffer(50mm tris-hcl,150mm nacl,5mm咪唑,8m尿素,ph 8.0)溶解,得到融合蛋白溶液。
48.(3)融合蛋白复性
49.配制4种不同的复性buffer,分别为:buffer1:1m tris-hcl,ph 7.8,5%甘油,2mm tcep;buffer2:500mm nacl,20mm tris-hcl,ph 9.5,2mm edta,5%甘油,2mm tcep;buffer3:0.2m arg,500mm nacl,20mm tris-hcl,ph 9.0,2mm edta,5%甘油,2mm tcep,0.01%tween-20;buffer4:0.2m arg,500mm nacl,20mm tris-hcl,ph 9.0,2mm edta,5%甘油,2mm tcep,0.01%tween-20,800mm尿素。
50.将上述4种复性buffer分别与所述融合蛋白进行透析复性:1)把2.5k透析袋剪成
适当长度10cm左右的小段。2)将透析袋放于在沸水中煮沸10分钟。3)取出透析袋,将水排出,用透析夹封住一端,往透析袋中加入步骤(2)所得融合蛋白溶液后排气泡并将两个末端均匀透析夹封住。4)将装有不同蛋白的透析袋放置于5l复性缓冲液中进行透析,每隔30min更换一次透析液,直至尿素透析干净,即得复性后的融合蛋白。
51.其中,本发明测试发现融合蛋白(融合多肽的包涵体蛋白)在高tris(1m tris)和高尿素(800mm尿素)的条件下不容易沉淀,但是尿素影响sumo蛋白酶的剪切效率,而高tris(1m)对sumo蛋白酶的剪切无影响。因此优选buffer1:1m tris-hcl,ph 7.8,5%甘油,2mm tcep。从而可以有效解决由于eddie具有较多的疏水性氨基酸,在透析除去尿素后易沉淀,不易复性的问题。
52.(4)采用sumo蛋白酶剪切去除融合蛋白中的sumo标签:
53.以步骤(3)复性后的融合蛋白为底物,加入sumo蛋白酶,底物浓度控制在1mg复性融合蛋白/ml复性缓冲液,30℃反应3.5h,得到酶切产物;同时,分别以未复性的相应蛋白为阴性对照分别(图4)。
54.其中,所述sumo蛋白酶的体积(ml)为待剪切复性融合蛋白的质量(mg)
×
0.007
÷
sumo蛋白酶浓度(mg/ml),所述sumo蛋白酶为thermofisher 12588018。
55.(5)酶切产物纯化:利用多肽的疏水作用,采用c8反向柱纯化来纯化多肽
56.将步骤(4)所得酶切产物5ml分2次加入到经100%甲醇活化的200mg/3ml的c8小柱上,依靠重力自流,标签蛋白不结合c8柱全部流出,而多肽能结合到c8柱上,待柱的末端无液体流出后用3ml的5%乙腈进行洗涤,依靠重力自流待洗涤液完全流尽,再利用2ml的95%乙腈将多肽洗脱下来,即得纯化后的目标多肽。
57.洗脱的多肽hplc检测,纯度较高,b147-1纯度为:93%,glp-1纯度为83%,cherry50纯度为:91%,cherry80纯度为:86%(图5),冻干后性状呈白色粉末状(图6)。可见,本发明方法不仅操作方便而且纯化得到的多肽纯度较好,并能够除盐,通用性高。
58.其中,所述c8反相柱的参数为碳含量:9%,比表面积:280m2/g粒径:40-75μm平均孔径:
59.综上,本发明结合eddie和sumo蛋白两种标签的优点,首次利用eddie(c69e、c168e)加sumo联合标签在c端融合目的多肽,并利用sumo蛋白酶切除标签,采用固相萃取小柱c8反相柱纯化酶切产物,获得多肽,以此建立大肠杆菌高效表达系统,从而得到高表达、高纯度的目的多肽,主要用于线性多肽的生物合成,具有产量高、成本低、易于纯化的优点,是一种生产多肽的新方法。本发明方法解决了生物表达过程小肽易降解以及满足了表达毒肽或抗菌肽的需求。
60.本发明提供了一种提高线性多肽生物表达的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。