1.本发明属于医药技术领域,具体涉及一种吴茱萸碱衍生物及其应用。
背景技术:
2.当前,癌症已经成为一种严重威胁人类健康和生命的疾病。目前,临床上主要采用国际通行的标准化疗方式治疗肿瘤,主要包括单一靶向药物治疗方式和多种药物联合用药,然而这些治疗方案普遍具有毒副作用大、患者依从性差等明显缺陷。而多靶点药物可以同时作用于疾病网络中的多个靶点,对各靶点的作用产生协同效应,使总效应大于各单效应之和,从而达到最佳的治疗效果。此外,单分子多靶点药物拥有相对简单的药代动力学性质,可以克服药物-药物相互作用产生的不良反应。多靶点药物已经成为新一代抗肿瘤药物研发的重要方向。
3.表观遗传学(epigenetics)与遗传学相对应,其主要包括非基因序列改变所致的基因表达水平变化,如染色质的构象变化和dna甲基化等,癌症的发生通常与表观遗传的失调密切相关。研究发现,表观遗传中的组蛋白乙酰化和去乙酰化调控异常将导致细胞染色质的重塑和空间构型发生变化,影响正常基因的表达,导致抑癌基因或与细胞周期调控相关蛋白出现异常,进而促进肿瘤的发生和转移。因此,对表观遗传相关靶点的调控已经成为抗肿瘤药物研发新方向,尤其是表观遗传学靶点组蛋白去乙酰化酶(histone deacetylases,hdacs)的研究已成为当前研究热点。
4.组蛋白的乙酰化/去乙酰化是染色体结构改变和基因表达的重要调节方式,在细胞凋亡、能量代谢、转录和翻译等生命过程中发挥重要作用。hdacs水解组蛋白中赖氨酸侧链上n-端乙酰基,使得核小体变得更加紧密,从而抑制基因的转录。它能调控细胞的多种功能和过程,例如基因表达、染色体改造、细胞增殖、分化、凋亡等。
5.典型的hdac抑制剂包含一个帽子结构(cap group,cap),一个锌离子结合区(zinc binding group,zbr),和一个合适的连接基团(linker)。研究表明,hdac与p53、热休克蛋白(hsp90)、拓扑异构酶(topisomerase,top)和微管蛋白(tubulin)等多个抗肿瘤靶点表现出抗肿瘤的协同效应,针对hdac和协同靶点的多靶点药物研究已经成为克服肿瘤耐药性,增强抗肿瘤疗效的有效手段,目前已有数个hdac多靶点抑制剂进入临床或临床前研究。
6.在前期研究中,发现新型top1抑制剂吴茱萸碱(evodiamine),对吴茱萸碱进行了系统的结构优化和构效关系研究,使其抗肿瘤活性显著提高,合成了一批吴茱萸碱类的衍生物,已经申请的专利如下:公开号为cn101787025a的专利申请公开了一种取代吴茱萸碱类抗肿瘤和抗真菌化合物及其制备方法;公开号为cn102311434a的专利申请公开了一种吴茱萸碱类化合物及其制备方法与应用;公开号为cn103992336a的专利申请公开了一种氧杂或硫杂吴茱萸碱类抗肿瘤衍生物及其制备方法。进一步地,通过深入的抗肿瘤作用机制研究发现,吴茱萸碱衍生物是top1和top2双重抑制剂,能够有效诱导肿瘤细胞凋亡,阻滞肿瘤细胞周期于g2/m期。其中,代表化合物3-氨基-10-羟基吴茱萸碱表现出优秀的体外和体内抗肿瘤活性。目前,top1/top2/hdac多靶点抑制剂的设计、合成和抗肿瘤活性研究未见文献
报道。
技术实现要素:
7.本发明的第一个目的是提供一种吴茱萸碱衍生物。
8.本发明的第二个目的是提供一种所述吴茱萸碱衍生物在制备抗肿瘤药物中的应用。
9.为了实现上述目的,本发明采用的技术方案如下:
10.本发明的第一方面提供了一种吴茱萸碱衍生物或其药用盐,具有以下结构通式:
[0011][0012]
其中:r选自氢、羟基、c1~c10烷基(优选c1~c5烷基)、c1~c10烷氧基(优选c1~c5烷氧基);
[0013]
x选自
[0014][0015]
n选自1至10的整数;
[0016]
a选自羟基、
[0017]
较优选的,所述吴茱萸碱衍生物中,
[0018]
r选自氢、羟基、甲基、乙基、正丙基、异丙基、甲氧基、乙氧基;
[0019]
x选自
[0020][0021]
n选自2、3、4、5、6、7;
[0022]
a选自羟基、
[0023]
最优选的,所述吴茱萸碱衍生物的结构选自以下结构的一种:
[0024]
[0025][0026]
本发明的第二方面提供了一种所述吴茱萸碱衍生物或其药用盐在制备抗肿瘤药物中的应用。
[0027]
所述肿瘤选自肠癌、肺癌、乳腺癌等。
[0028]
所述药用盐是其有机酸盐或无机酸盐;所述无机酸为盐酸、硫酸、磷酸、二磷酸、氢溴酸或硝酸;所述有机酸为乙酸、马来酸、富马酸、酒石酸、琥珀酸、乳酸、对甲苯磺酸、水杨酸、草酸、鞣酸、枸橼酸、三氟醋酸、苹果酸或苯磺酸盐。
[0029]
所述药用盐不含结晶水,或含一个或一个以上结晶水。
[0030]
本发明的第三方面提供了一种所述吴茱萸碱衍生物或其药用盐在制备hdac抑制剂、top1抑制剂、top2抑制剂或hdac、top1和top2三靶点抑制剂中的应用。
[0031]
所述吴茱萸碱衍生物作为hdac、top1和top2三靶点抑制剂用于治疗恶性肿瘤或与
分化增值相关疾病的药物。
[0032]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0033]
本发明提供了基于拓扑异构酶1(top1)、拓扑异构酶2(top2)和组蛋白去乙酰化酶(hdac)多靶点的小分子抗癌药物,药理实验表明,本发明所述吴茱萸碱衍生物对拓扑异构酶1、拓扑异构酶2和组蛋白去乙酰化酶均具有很强的抑制活性,而且具有较强的体内外抗肿瘤活性,因此,可以作为拓扑异构酶抑制剂、组蛋白去乙酰化酶抑制剂和抗肿瘤的药物使用。
[0034]
本发明提供的吴茱萸碱衍生物,其中化合物29b和45b能同时抑制top1、top2和hdac,在hct116细胞系上显示出良好的体外抗肿瘤活性。在hct116细胞中,它们有效诱导g2细胞周期阻滞的凋亡。在人结肠癌hct-116裸鼠移植瘤实验中,化合物29b和45b具有优秀的抗肿瘤药效,抑瘤率高达59.02%(29b)和69.63%(45b),显著优于阳性对照药组saha。总之,这项工作为基于吴茱萸碱的top/hdac双靶抑制剂提供了有价值的sar信息和先导化合物,并指导进一步的结构优化和抗肿瘤药物的发现。
附图说明
[0035]
图1是吴茱萸碱衍生物对top1和top2抑制实验结果示意图,其中,a是吴茱萸碱衍生物在浓度为200μm时对top1的抑制实验结果示意图,b是吴茱萸碱衍生物在浓度为100μm时对top1的抑制实验结果示意图,c是吴茱萸碱衍生物在浓度为50μm时对top1的抑制实验结果示意图,条带1,超螺旋质粒dna pbr322;条带2,dna top1;条带3,dna top1 cpt;条带4-20,dna top1 目标化合物(23a,23b,23c,16a,17a,18a,16b,17b,18b,16c,17c,18c,24a,24b,24c,29a,29b)/(37d,38d,36d,37c,38c,36c,37b,38b,36b,37a,38a,36a,45a,45b,45c,29c);d是吴茱萸碱衍生物在浓度为300μm时对top2的抑制实验结果示意图,e是吴茱萸碱衍生物在浓度为200μm时对top2的抑制实验结果示意图,f是吴茱萸碱衍生物在浓度为100μm时对top2的抑制实验结果示意图,条带1,超螺旋质粒dnapbr322;条带2,dna top1;条带3,dna top1 eto;条带4-16,dna top1 目标化合物(23a,23b,23c,29a,29b,29c,37a,37b,37c,37d,45a,45b,45c);g是eto和吴茱萸碱衍生物29b以及45b分别在浓度为300μm、200μm、100μm以及50μm时对top2的抑制实验结果示意图。
[0036]
图2是吴茱萸碱衍生物体内抗肿瘤效果的结果示意图,其中,a是时间与肿瘤体积的结果示意图,b是肿瘤重量的实验结果示意图,c是给药结束后肿瘤照片结果示意图,d是时间与重量的实验结果示意图。
具体实施方式
[0037]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0038]
下述实施例中未注明具体条件的实验方法,通常按照常规条件或制造厂商所建议的条件进行。实施例中所述的百分比除特别说明外,均为重量百分比。
[0039]
化合物16-18的合成
[0040][0041]
reagents and conditions:(a)hbtu,et3n,mecn,2h,rt,yield 45%-50%;(b)pcc,dcm,2h,rt,yield 54-76%;(c)nabh3cn,meoh,3h,rt,yield 50-62%;(d)ch3oh,nh2oh
·
hcl,koh,40℃,45min,yield 82-89%.
[0042]
实施例1
[0043]n1-羟基-n
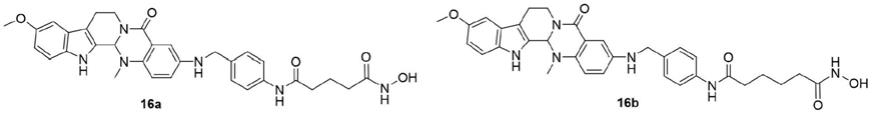
5-(4-((10-甲氧基-14-甲基-5-氧代-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)苯基)戊二酰胺化合物16a的制备
[0044]
(1)制备中间体11a:甲基-5-((4-(羟甲基)苯基)氨基)-5-氧代戊酸甲酯
[0045]
将化合物9(1.0g,8.12mmol)、戊二酸单甲酯10a(1.3g,8.93mmol)、hbtu(4.6g,12.18mmol)和三乙胺(5.0ml)溶解在15ml dmf中,并在室温下搅拌2h。反应结束后,将反应溶液在150ml水中稀释,并用50ml乙酸乙酯萃取三次。然后用mgso4干燥合并的有机层,通过硅胶柱色谱(石油醚:乙酸乙酯=2:1)纯化,得到0.78g黄色固体中间体11a,产率36%。1h-nmr(300mhz,dmso-d6)δ:9.85(s,1h),7.51(d,j=8.5hz,2h),7.21(d,j=8.4hz,2h),5.08(s,1h),4.50-4.32(m,2h),3.59(s,3h),2.40-2.29(m,4h),1.82(m,2h).
[0046]
(2)制备中间体12a:甲基-5-((4-甲酰基苯基)氨基)-5-氧代戊酸酯
[0047]
将化合物11a(0.8g,3.18mmol)溶于50ml二氯甲烷中,在常温下滴加100ml溶有pcc(1.36g,3.72mmol)的二氯甲烷溶液,室温下搅拌2小时,tlc点板监测,待反应结束后,利用硅藻土除去pcc,之后将反应液减压蒸干,柱层析(pe:ea=1:1)得0.59g白色固体中间体12a,产率为76%。1h-nmr(300mhz,dmso-d6)δ:10.34(s,1h),9.86(s,1h),7.82(dd,j=8.8hz,4h),3.59(s,3h),2.42(t,j=5.8hz,2h),2.40-2.34(t,2h),1.92-1.82(m,2h).
[0048]
(3)制备中间体13a:甲基5-((4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)苯基)氨基)-5-氧代戊二酸酯
[0049]
化合物7a的结构如下:
[0050][0051]
将中间体12a(0.14g,0.56mmol)和化合物7a(0.16g,0.6mmol)溶于25ml甲醇中,滴加两滴乙酸,于室温下搅拌30分钟后加入nabh3cn(0.08g,1.29mmol),于室温下反应3小时,tlc点板监测,待反应结束后浓缩,用洗脱剂(ch2cl2:meoh=100:5)进行柱层析得0.122g黄色固体中间体13a,产率为50%。1h-nmr(600mhz,dmso-d6)δ:11.07(s,1h),9.86(s,1h),7.52(d,j=8.5hz,2h),7.28(d,j=8.6hz,2h),7.24(d,j=8.7hz,1h),7.07(d,j=2.8hz,1h),7.00(d,j=2.5hz,1h),6.97(d,j=8.6hz,1h),6.81(dd,j=8.7,2.8hz,1h),6.75(dd,j=8.8,2.5hz,1h),6.29(t,j=6.0hz,1h),5.87(s,1h),4.67-4.61(m,1h),4.21(d,j=5.9hz,2h),3.76(s,3h),3.59(s,3h),3.14-3.06(m,1h),2.89-2.69(m,2h),2.36(t,j=7.4hz,2h),2.33(t,j=7.4hz,2h),2.27(s,3h),1.88-1.81(m,2h).
[0052]
(4)制备目标化合物16a
[0053]
称取盐酸羟胺(4.67g,67mmol)溶于24ml甲醇中,并于冰浴下搅拌,加入氢氧化钾(5.61g,100mmol)溶于甲醇(14ml)的溶液,保持零摄氏度搅拌30分钟,反应结束后将其过滤得新鲜制备的羟胺甲醇溶液。将中间体13a(0.12g,0.2mmol)溶于上述新鲜制备的羟胺甲醇溶液(15ml)中,于室温下反应2h,待反应结束后,用稀盐酸将反应ph调至7析出固体,将其过滤,水洗得0.104g黄色固体即化合物16a,产率为89%。1h-nmr(600mhz,dmso-d6)δ:11.07(s,1h),10.40(s,1h),9.87(s,1h),8.71(s,1h),7.58-7.51(m,2h),7.29(d,j=8.2hz,2h),7.26(d,j=8.7hz,1h),7.08(d,j=2.8hz,1h),7.01(d,j=2.5hz,1h),6.98(d,j=8.6hz,1h),6.83(dd,j=8.7,2.8hz,1h),6.77(dd,j=8.7,2.5hz,1h),6.29(t,j=6.0hz,1h),5.88(s,1h),4.70-4.58(m,1h),4.23(d,j=5.8hz,2h),3.77(s,3h),3.16-3.09(m,j=12.3,4.1hz,1h),2.89-2.71(m,2h),2.30(d,j=13.5hz,5h),2.02(t,j=7.5hz,2h),1.84-1.79(m,2h).
13
c-nmr(150mhz,dmso-d6)δ:171.06,169.17,164.38,153.75,145.67,140.75,138.35,134.98,132.34,130.46,127.84,126.42,124.24,123.79,119.58,118.69,112.68,112.34,111.73,110.13,100.68,69.44,55.82,46.89,39.61,37.44,36.04,32.07,21.63,20.39.hrms(esi
):m/z calcd for[m h]
c32h35
n6o5:583.2664,found:583.2679.
[0054]
实施例2
[0055]
化合物16b的制备:将实施例1中第一步化合物10a替换为化合物10b(n=4,m=1),其他同实施例1,制备得到化合物16b,产率为87%。
[0056]
实施例3
[0057]
化合物16c的制备:将实施例1中第一步化合物10a替换为化合物10c(n=5,m=2),其他同实施例1,制备得到化合物16c,产率为85%。
[0058]
实施例4
[0059]
化合物17a的制备:将实施例1中第三步化合物7a替换为化合物7b,其他同实施例1,制备得到化合物17a,产率为82%。
[0060]
化合物7b的结构如下:
[0061][0062]
实施例5
[0063]
化合物17b的制备:将实施例4中第一步化合物10a替换为化合物10b(n=4,m=1),第三步化合物7a替换为化合物7b,其他同实施例4,制备得到化合物17b,产率为86%。
[0064]
实施例6
[0065]
化合物17c的制备:将实施例4中第一步化合物10a替换为化合物10c(n=5,m=2),第三步化合物7a替换为化合物7b,其他同实施例4,制备得到化合物17c,产率为85%。
[0066]
实施例7
[0067]
化合物18a的制备:将实施例1中第三步化合物7a替换为化合物7c,其他同实施例1,制备得到化合物18a,产率为81%。
[0068]
化合物7c的结构如下:
[0069][0070]
实施例8
[0071]
化合物18b的制备:将实施例7中第一步化合物10a替换为化合物10b(n=4,m=1),第三步化合物7a替换为化合物7c,其他同实施例7,制备得到化合物18b,产率为84%。
[0072]
实施例9
[0073]
化合物18c的制备:将实施例7中第一步化合物10a替换为化合物10c(n=5,m=2),第三步化合物7a替换为化合物7c,其他同实施例7,制备得到化合物18c,产率为87%。
[0074]
化合物23a-c和24a-c的合成
[0075][0076]
reagent and conditions:(a)nabh3cn,meoh,3h,rt,yield 50-62%;(b)thf/h2o/ch3oh=
[0077]
1:1:1,lioh,4h,rt,yield 82%;(c)ch3oh,nh2oh
·
hcl,koh,40℃,45min,yield 82-89%;(d)dmf,dipea,hatu,3h,rt,yield 46-52%.
[0078]
实施例10
[0079]
n-羟基-4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六氢吲哚[2',3':3,
4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)苯基)苯甲酰胺即化合物23a的制备
[0080]
(1)制备中间体21a:甲基4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶[2,1-b]喹唑啉-3-基)氨基)苯甲酸甲酯
[0081]
将化合物19(0.56mmol)和化合物7a(0.6mmol)按照实施例1制备中间体13a的方法步骤操作,柱层析得到180mg黄色固体中间体21a,收率64%。1h-nmr(600mhz,dmso-d6)δ:11.05(s,1h),7.99-7.85(m,2h),7.51(d,j=8.1hz,2h),7.24(d,j=8.7hz,1h),7.04(d,j=2.8hz,1h),7.00(d,j=2.5hz,1h),6.98(d,j=8.6hz,1h),6.81(dd,j=2.8,8.6hz,1h),6.75(dd,j=2.4,8.8hz,1h),6.45(t,j=6.1hz,1h),5.87(s,j=1.5hz,1h),4.65-4.58(m,1h),4.38(d,j=6.1hz,2h),3.83(s,3h),3.76(s,3h),3.12-3.05(m,1h),2.87-2.67(m,2h),2.28(s,3h).
[0082]
(2)制备目标产物化合物23a
[0083]
中间体21a(0.2mmol)溶于上述新鲜制备的羟胺甲醇溶液(15ml)中,其他方法步按照实施例1中化合物16a的方法步骤操作,柱层析得到104mg黄色固体化合物23a,收率89%。1h-nmr(600mhz,dmso-d6)δ:11.07(s,1h),10.40(s,1h),9.87(s,1h),8.71(s,1h),7.58-7.51(m,2h),7.29(d,j=8.2hz,2h),7.26(d,j=8.7hz,1h),7.08(d,j=2.8hz,1h),7.01(d,j=2.5hz,1h),6.98(d,j=8.6hz,1h),6.83(dd,j=8.7,2.8hz,1h),6.77(dd,j=8.7,2.5hz,1h),6.29(t,j=6.0hz,1h),5.88(s,1h),4.70-4.58(m,1h),4.23(d,j=5.8hz,2h),3.77(s,3h),3.16-3.09(m,j=12.3,4.1hz,1h),2.89-2.71(m,2h),2.30(d,j=13.5hz,5h),2.02(t,j=7.5hz,2h),1.84-1.79(m,2h).
13
c-nmr(150mhz,dmso-d6)δ:171.06,169.17,164.38,153.75,145.67,140.75,138.35,134.98,132.34,130.46,127.84,126.42,124.24,123.79,119.58,118.69,112.68,112.34,111.73,110.13,100.68,69.44,55.82,46.89,39.61,37.44,36.04,32.07,21.63,20.39.hrms(esi
):m/z calcd for[m h]
c32h35
n6o5:583.2664,found:583.2679.
[0084]
实施例11
[0085]
化合物23b的制备:将实施例10中第一步化合物7a替换为化合物7b,其他同实施例10,制备得到化合物23b,产率为87%。
[0086]
实施例12
[0087]
化合物23c的制备:将实施例10中第一步化合物7a替换为化合物7c,其他同实施例10,制备得到化合物23c,产率为81%。
[0088]
实施例13
[0089]
n-(2-氨基苯基)-4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)苯甲酰胺即化合物24a的制备
[0090]
将化合物21a(0.1g,0.2mmol)溶解在15ml的meoh/thf/h2o(1:1:1)的混合溶液中,并添加lioh(0.03g,1.2mmol)。在室温下搅拌2小时后,用乙酸将溶液ph调节至7。将混合物浓缩,得到黄色固体化合物22a(0.082g,85%)。粗产物用于下一步,无需进一步纯化。
[0091]
将化合物22a(0.12g,0.24mmol)、邻苯二胺(0.053g,0.48mmol)、hatu(0.15g,0.39mmol)和dipea(91μl,0.52mmol)加入到干燥的dmf(10ml)中,并在室温下搅拌2h。然后加入100ml水,并用乙酸乙酯(50ml
×
3)萃取。用无水mgso4干燥,合并有机层,柱层析(石油醚:乙酸乙酯=1:1)分离,得到0.057g黄色固体化合物24a,产率46%。1h-nmr(600mhz,
dmso-d6)δ:11.07(s,1h),9.62(s,1h),7.94(d,j=7.9hz,2h),7.49(d,j=8.0hz,2h),7.24(d,j=8.8hz,1h),7.15(d,j=7.8hz,1h),7.06(d,j=2.7hz,1h),6.96(dd,j=8.2,11.2hz,3h),6.83(dd,j=2.8,8.6hz,1h),6.82-6.74(m,2h),6.58(t,j=7.5hz,1h),6.49(t,j=6.2hz,1h),5.87(s,1h),4.89(s,2h),4.62(dd,j=4.5,12.5hz,1h),4.38(d,j=6.0hz,2h),3.76(s,3h),3.20-3.00(m,1h),2.82-2.76(m,2h),2.28(s,3h).
13
c-nmr(150mhz,dmso-d6)δ:165.63,164.34,153.75,145.42,144.40,143.56,140.88,133.55,132.32,130.48,128.35,127.23,127.10,126.87,126.42,124.20,123.83,118.76,116.69,116.56,112.69,112.35,111.72,110.16,100.65,69.45,55.81,46.91,37.47,20.38.hrms(esi
):m/z calcd for[m h]
c34h33
n6o3:573.2609,found:573.2609.
[0092]
实施例14
[0093]
化合物24b的制备:将实施例13中化合物21a替换为化合物21b,其他同实施例13,制备得到化合物24b,产率为47%。
[0094]
实施例15
[0095]
化合物24c的制备:将实施例13中化合物21a替换为化合物21c,其他同实施例13,制备得到化合物24c,产率为42%。
[0096]
化合物29a-c的合成
[0097][0098]
reagent and conditions:(a)nabh3cn,meoh,3h,rt,yield 52-64%;(b)thf/h2o/ch3oh,lioh,4h,rt,yield 80-84%;(c)dmf,dipea,hatu,3h,rt,yield 48-52%.(d)bbr3,et2o,dcm,2h,rt,yield 74-76%.
[0099]
实施例16
[0100]
(e)-n-羟基-3-(4-((10-甲氧基-14-甲基-5-氧代-5,7,8,13,13b,14-六氢吲哚并[2',3':3,4]吡啶并[2,1-b]喹唑啉-3-基)氨基)甲基)苯基)丙烯酰胺即化合物29a的制备
[0101]
(1)制备中间体26a:甲基(e)-3-(4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)丙烯酸甲酯
[0102]
将化合物25(0.56mmol)和化合物7a(0.6mmol)溶于25ml甲醇中,其他方法步骤与实施例1中制备中间体13a相同,柱层析得到120mg黄色固体中间体26a,收率81%。1h-nmr
(600mhz,dmso-d6)δ:11.05(s,1h),7.68(d,j=8.2hz,2h),7.64(d,j=16.0hz,1h),7.41(d,j=8.2hz,2h),7.24(d,j=8.8hz,1h),7.06(d,j=2.8hz,1h),7.00(d,j=2.5hz,1h),6.97(d,j=8.6hz,1h),6.82(dd,j=2.8,8.7hz,1h),6.75(dd,j=2.4,8.8hz,1h),6.60(d,j=16.0hz,1h),6.40(t,j=6.1hz,1h),5.86(s,j=1.4hz,1h),4.62-4.60(m,1h),4.32(d,j=5.9hz,2h),3.76(s,3h),3.72(s,3h),3.15-3.05(m,1h),2.89-2.67(m,2h),2.28(s,3h).
[0103]
(2)制备目标产物化合物29a
[0104]
将中间体26a(0.2g,0.4mmol)溶解在meoh/thf/h2o(1:1:1)混合溶液(15ml)中,然后加入lioh(0.06g,2.4mmol)。将混合物在室温下搅拌2小时,反应结束后用乙酸调节至ph为7,析出固体后抽滤,得到黄色固体化合物27a(0.164g,85%),无需进一步纯化。
[0105]
将化合物27a(0.164g,0.32mmol)、邻三苯基羟胺(0.10g,0.38mmol)、hatu(0.16g,0.38mmol)和dipea(106μl,0.64mmol)溶解于dmf(10ml)中。将混合物在室温下搅拌2h。反应后,加入饱和氯化钠溶液(100ml)并用乙酸乙酯(50ml
×
3)萃取。用无水mgso4干燥,合并有机层,减压浓缩并通过柱层析(石油醚:乙酸乙酯=2:1)分离,得到黄色固体化合物28a(0.126g,产率51%)。
[0106]
将化合物28a(0.126g,0.164mmol)溶解在ch2cl2(10ml)中,然后加入bbr3(0.3ml),并在室温下搅拌混合物2h。反应结束后,加入10ml水使反应停止,过滤沉淀产物,用水冲洗,干燥后得0.086g黄色固体化合物29a,产率74%。1h-nmr(600mhz,dmso-d6)δ:11.08(s,1h),7.53(d,j=7.9hz,2h),7.45(d,j=15.8hz,1h),7.41(d,j=7.9hz,2h),7.26(d,j=8.7hz,1h),7.09(d,j=2.8hz,1h),7.01(d,j=2.4hz,1h),6.99(d,j=8.6hz,1h),6.83(dd,j=2.8,8.6hz,1h),6.77(dd,j=2.5,8.8hz,1h),6.46(d,j=15.8hz,1h),6.41(d,j=6.2hz,1h),5.88(s,1h),4.64(m,1h),4.32(d,j=5.8hz,2h),3.77(s,3h),3.11(m,1h),2.91-2.69(m,2h),2.29(s,3h).
13
c-nmr(150mhz,dmso-d6)δ:164.37,163.23,153.77,145.54,142.29,140.86,138.47,133.84,132.35,130.48,128.05,128.01,126.44,124.25,123.85,119.04,118.68,112.71,112.36,111.75,110.14,100.68,69.45,55.83,49.08,47.00,37.46,20.40.hrms(esi
):m/z calcd for[m h]
c30h30
n5o4:524.2292,found:524.2293.
[0107]
实施例17
[0108]
化合物29b的制备:将实施例16中第一步化合物7a替换为化合物7b,其他同实施例16,制备得到化合物29b,产率为72%。
[0109]
实施例18
[0110]
化合物29c的制备:将实施例16中第一步化合物7a替换为化合物7c,其他同实施例16,制备得到化合物29c,产率为68%。
[0111]
化合物36-38的合成
[0112][0113]
reagents and conditions:(a)k2co3,dmf,4h,rt,yield 78-85%;(b)nabh3cn,meoh,3h,rt,yield 52-64%;(c)ch3oh,nh2oh
·
hcl,koh,40℃,45min,yield 78-88%.
[0114]
实施例19
[0115]
n-羟基-5-(4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六羟基吲哚[2',3':3,4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)苯氧基)戊酰胺即化合物36a的制备
[0116]
(1)制备中间体32a:5-(4-甲酰苯氧基)戊酸乙酯
[0117]
取对羟基苯甲醛即化合物30(2g,16.38mmol)于100ml圆底烧瓶中,加入20ml dmf,再加入碳酸钾(2.5g,18.02mmol),室温下反应10分钟,之后加入化合物31a(3.5g,16.70mmol),在65℃下反应2小时。反应结束后,加入饱和食盐水(200ml),乙酸乙酯(50ml
×
3)萃取。合并有机层,无水硫酸镁干燥,抽滤,减压蒸干溶剂。剩余物用柱色谱纯化,流动相为石油醚/乙酸乙酯混合溶剂(100:2),得3.48g黄色油状物中间体32a,收率为85%。1h-nmr(600mhz,dmso-d6)δ:9.87(s,1h),7.87(d,j=6.0hz,2h),7.13(d,j=6.0hz,2h),4.11(t,j=6.8hz,2h),4.06(m,2h),2.38(t,j=7.3hz,2h),1.80-1.74(m,2h),1.73-1.66(m,2h),1.18(t,j=7.1hz,3h).
[0118]
(2)制备中间体33a:6-(4-((10-甲氧基-14-甲基-5-氧代-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶[2,1-b]喹唑啉-3-基)氨基)甲基)苯氧基)己酸乙酯
[0119]
将中间体33a(0.56mmol)和化合物7a(0.6mmol)溶于25ml甲醇中,其他方法步骤与实施例1中制备中间体13a相同,柱层析得到450mg黄色固体中间体33a,收率71%。1h-nmr(600mhz,dmso-d6)δ:11.07(s,1h),7.28(d,j=8.6hz,2h),7.25(d,j=8.7hz,1h),7.09(d,j=2.8hz,1h),7.01(d,j=2.5hz,1h),6.98(d,j=8.6hz,1h),6.88(d,j=8.7hz,2h),6.83(dd,j=8.7,2.8hz,1h),6.76(dd,j=8.7,2.5hz,1h),6.25(t,j=6.0hz,1h),5.87(s,1h),4.64(m,1h),4.21(d,j=5.9hz,2h),4.09-4.02(m,2h),3.94(t,j=6.0hz,2h),3.77(s,3h),3.19-3.05(m,1h),2.93-2.68(m,2h),2.36(t,j=7.2hz,2h),2.28(s,3h),1.79-1.57(m,4h),1.18(t,j=7.1hz,3h).
[0120]
(3)制备目标产物36a
[0121]
中间体33a(0.2mmol)溶于上述新鲜制备的羟胺甲醇溶液(15ml)中,其他方法步按照实施例1中化合物16a的方法步骤操作,柱层析得到120mg黄色固体即化合物36a,收率43%。1h-nmr(600mhz,dmso-d6)δ:11.07(s,1h),10.35(s,1h),8.68(s,1h),7.28(d,j=
5.7hz,2h),7.26(d,j=5.6hz,1h),7.10(d,j=2.8hz,1h),7.01(d,j=2.5hz,1h),6.98(d,j=8.6hz,1h),6.88(d,j=8.6hz,1h),6.83(dd,j=2.8,8.6hz,1h),6.77(dd,j=2.5,8.7hz,1h),6.25(t,j=6.0hz,1h),5.88(s,j=1.4hz,1h),4.68-4.57(m,1h),4.20(d,j=5.8hz,2h),3.92(t,j=6.3hz,2h),3.77(s,3h),3.17-3.05(m,1h),2.91-2.70(m,2h),2.29(s,3h),1.96(t,j=7.2hz,2h),1.71-1.58(m,4h).
13
c-nmr(150mhz,dmso-d6)δ:168.55,164.41,158.03,153.76,145.76,140.74,132.40,132.14,130.50,128.80,126.44,124.26,123.79,118.66,114.76,112.72,112.33,111.72,110.12,100.70,69.47,67.64,55.85,46.75,37.45,32.87,28.88,22.66,20.42.hrms(esi-):m/z calcd for[m-h]-c
32h34
n5o5:568.2565,found:568.2556。
[0122]
实施例20
[0123]
化合物36b的制备:将实施例19中的化合物31a替换为化合物31b,其他同实施例19,产率为44%。
[0124]
实施例21
[0125]
化合物36c的制备:将实施例19中的化合物31a替换为化合物31c,其他同实施例19,产率为54%。
[0126]
实施例22
[0127]
化合物36d的制备:将实施例19中的化合物31a替换为化合物31d,其他同实施例19,产率为46%。
[0128]
实施例23
[0129]
化合物37a的制备:将实施例19中的化合物7a替换为化合物7b,其他同实施例19,产率为48%。
[0130]
实施例24
[0131]
化合物37b的制备:将实施例23中的化合物31a替换为化合物31b,其他同实施例23,产率为47%。
[0132]
实施例25
[0133]
化合物37c的制备:将实施例23中的化合物31a替换为化合物31c,其他同实施例23,产率为39%。
[0134]
实施例26
[0135]
化合物37d的制备:将实施例23中的化合物31a替换为化合物31d,其他同实施例23,产率为55%。
[0136]
实施例27
[0137]
化合物38a的制备:将实施例19中的化合物7a替换为化合物7c,其他同实施例19,产率为68%。
[0138]
实施例28
[0139]
化合物38b的制备:将实施例27中的化合物31a替换为化合物31b,其他同实施例27,产率为49%。
[0140]
实施例29
[0141]
化合物38c的制备:将实施例19中的化合物31a替换为化合物31c,其他同实施例27,产率为53%。
[0142]
实施例30
[0143]
化合物38d的制备:将实施例19中的化合物31a替换为化合物31d,其他同实施例27,产率为54%。
[0144][0145][0146]
reagents and conditions:(a)na2co3,ch3ch2oh,pd/c,h2,48h,rt,yield 50%;(b)mcpba,dcm,na2s2o3,nahco3,rt,2h,yield 75%;(c)k2co3,dmf/mecn,1h,rt,yield80%;(d)pcc,dcm,2h,rt,yield 56-74%;(e)nabh3cn,meoh,3h,rt,yield 48-66%;(f)ch3oh,nh2oh
·
hcl,koh,40℃,45min,yield 78-88%.
[0147]
实施例31
[0148]
n-羟基-2-(4-((10-甲氧基-14-甲基-5-氧代-5,7,8,13,13b,14-六氢吲哚并[2',3':3,4]吡啶并[2,1-b]喹唑啉-3-基)氨基)甲基)哌啶-1-基)嘧啶-5-甲酰胺即化合物45a的制备
[0149]
(1)制备中间体40:2-(甲硫基)嘧啶-5-羧酸乙酯
[0150]
室温下将化合物39(0.2g,0.86mmol)溶解于30ml乙醇中,然后加入na2co3(0.091g)和10%的钯碳,在氢气下反应48h。然后,用硅藻土过滤混合物,并在减压下蒸发溶液。通过硅胶柱层析(石油醚:乙酸乙酯=100:2)纯化残余物,得到0.12g白色固体中间体40,产率为72%。1h-nmr(600mhz,dmso-d6)δ:9.00(s,2h),4.37-4.31(m,2h),2.57(s,3h),1.32(t,j=7.1hz,3h).
[0151]
(2)制备中间体41:2-(甲磺酰)嘧啶-5-羧酸乙酯
[0152]
在室温下将中间体40(0.424g,2.14mmol)溶解在含有3-氯过氧苯甲酸(2.0g,11.59mmol)的二氯甲烷溶液100ml中,反应2h。反应后,用na2s2o3溶液将其淬灭,用饱和nahco3水溶液洗涤,用mgso4干燥,过滤并浓缩得到0.374g白色固体中间体41,产率为76%。1h-nmr(600mhz,dmso-d6)δ:9.47(s,2h),4.46-4.41(m,2h),3.47(s,3h),1.37(t,j=7.1hz,3h).
[0153]
(3)制备中间体42:2-(4-(羟甲基)哌啶-1-基)嘧啶-5-羧酸乙酯
[0154]
将哌啶-4-甲醇(2.48g,21.55mmol)和k2co3(8.9g,64.65mmol)加入到dmf/mecn(1:1,20ml)混合溶液中,搅拌10分钟。然后加入中间体41(5g,21.55mmol)并搅拌30分钟。反应
结束后,用水(100ml)稀释混合物并用乙酸乙酯(50ml
×
3)萃取。将合并的有机层用无水mgso4干燥,通过硅胶柱色谱(二氯甲烷:甲醇=100:3)分离,得到3.68g橙色固体中间体42,产率为64%。1h-nmr(600mhz,dmso-d6)δ:8.75(s,2h),4.76(m,j=12.3,2.8hz,2h),4.50(t,j=5.3hz,1h),4.29-4.24(m,j=7.1hz,2h),3.27(t,j=5.7hz,2h),2.32-2.95(m,2h),1.78-1.65(m,3h),1.28(t,j=7.1hz,3h),1.12-1.04(m,2h).
[0155]
(4)制备中间体43:2-(4-甲酰哌啶-1-基)嘧啶-5-羧酸乙酯
[0156]
将中间体42(3.7g,14.05mmol)溶于50ml二氯甲烷中,其他方法步按照实施例1中化合物12a的方法步骤操作,柱层析得到2.32g黄色固体中间体43,收率65%。1h-nmr(600mhz,dmso-d6)δ:9.63(s,1h),8.78(s,2h),4.54(t,j=4.3hz,1h),4.51(t,j=4.3hz,1h),4.29-4.23(m,2h),3.34-3.31(m,2h),2.71-2.65(m,1h),1.95(dd,j=4.0,13.5hz,2h),1.53-1.45(m,2h),1.30(t,j=7.1hz,3h).
[0157]
(5)制备中间体44a:2-(4-((10-甲氧基-14-甲基-5-氧基-5,7,8,13,13b,14-六氢吲哚[2',3':3,4]吡啶基[2,1-b]喹唑啉-3-基)氨基)甲基)哌啶-1-基)嘧啶-5-羧酸乙酯
[0158]
将中间体43(0.56mmol)和化合物7a(0.6mmol)溶于25ml甲醇中,其他方法步骤与实施例1中制备中间体13a相同,柱层析得到160mg黄色固体中间体44a,收率45%。1h-nmr(600mhz,dmso-d6)δ:11.09(s,1h),8.76(s,2h),7.24(d,j=9.3hz,1h),7.07(d,j=2.8hz,1h),7.01(d,j=2.5hz,1h),6.99(d,j=8.6hz,1h),6.83(dd,j=8.6,2.8hz,1h),6.75(dd,j=8.8,2.5hz,1h),5.88(s,1h),5.79(t,j=5.8hz,1h),4.79(d,j=13.4hz,2h),4.70-4.60(m,1h),4.30-4.22(m,2h),3.76(s,3h),3.18-3.12(m,1h),3.04-2.93(m,4h),2.90-2.69(m,2h),2.28(s,3h),1.95-1.85(m,3h),1.28(t,j=7.1hz,3h),1.20-1.27(m,2h).
[0159]
(6)制备目标产物45a
[0160]
中间体44a(0.2mmol)溶于上述新鲜制备的羟胺甲醇溶液(15ml)中,其他方法步按照实施例1中化合物16a的方法步骤操作,柱层析得到130mg黄色固体化合物45a,收率67%。1h-nmr(600mhz,dmso-d6)δ:11.09(s,2h),9.01(s,1h),8.67(s,2h),7.26(d,j=8.7hz,1h),7.09(d,j=2.8hz,1h),7.02(d,j=2.5hz,1h),7.00(d,j=8.6hz,1h),6.84(dd,j=8.6,2.8hz,1h),6.77(dd,j=8.8,2.4hz,1h),5.89(s,1h),5.78(t,j=5.6hz,1h),4.80-4.73(m,2h),4.67(m,1h),3.77(s,3h),3.19-3.07(m,1h),3.00-2.92(m,4h),2.91-2.72(m,2h),2.30(s,3h),1.89(t,j=14.7hz,3h),1.20-1.63(m,2h).
13
c-nmr(150mhz,dmso-d6)δ:164.47,162.47,161.75,157.56,153.75,146.15,140.43,132.34,130.49,126.43,124.36,123.98,118.35,114.54,112.69,112.36,111.74,109.44,100.66,69.49,55.82,49.31,43.91,37.51,35.77,30.16,20.43.hrms(esi
):m/z calcd for[m h]
c31h35
n8o4:583.2776,found:583.2778.
[0161]
实施例32
[0162]
化合物45b的制备:将实施例31中的化合物7a替换为化合物7b,其他同实施例31,产率为71%。
[0163]
实施例33
[0164]
化合物45c的制备:将实施例31中的化合物7a替换为化合物7c,其他同实施例31,产率为65%。
[0165]
实施例1~33制备的化合物的结构式和核磁质谱数据如表1所示:
[0166]
表1.本发明优选化合物的结构式和核磁质谱数据
[0167]
[0168]
[0169]
[0170]
[0171]
[0172]
[0173][0174]
实施例34
[0175]
目标化合物酶抑制活性和体外抗肿瘤活性
[0176]
1.目标化合物hdac1酶和hdac2酶抑制测试
[0177]
1.1实验材料:
[0178]
hdac1酶(或hdac2酶),缓冲液(137mm氯化钠,2.7mm氯化钾,1mm氯化镁,0.1mg/mlbsa,ph=8的tris-hcl 25mm),hdac substrate 3,胰蛋白酶,96孔黑色板。
[0179]
1.2实验方法:
[0180]
(1)96孔黑色板平衡至室温;
[0181]
(2)用含有10%的dmso的缓冲液稀释待测化合物,化合物的浓度依次为100μm,30μm,10μm,3μm,1μm,0.3μm,0.1μm,0.03μm,0.01μm,0.003μm;
[0182]
(3)将11μl的hdac1(或hdac2酶)加入到400μl的缓冲液中,摇匀;
[0183]
(4)向96孔板上第2-11孔加入35μl刚刚配好的含有hdac1酶(或hdac2酶)的缓冲液,并依次加入5μl稀释好的不同浓度的化合物到对应的反应孔中,对于阴性对照(第一个孔)和空白对照孔(第十一个孔),分别加入40μl和5μl assay buffer;
[0184]
(5)向所有反应孔中加入100μm的hdac substrate 5μl和0.5mg/ml的胰蛋白酶5μl,37℃孵化30分钟后读数。
[0185]
(6)依据公式计算抑制率:抑制率=(100%活性孔-样品孔)/100%活性孔*100,在graphpad软件中将酶活性对化合物浓度的曲线进行拟合,求出化合物的ic
50
值;
[0186]
实验结果表明这些化合物都表现出良好的hdac1抑制活性,其中两个化合物29b(ic
50
=0.018μm)和45b(ic
50
=0.004μm)表现出优于阳性对照药saha(伏立诺他)(ic
50
=0.023μm)的hdac1抑制活性。
[0187]
表2.目标化合物hdac1和hdac2抑制活性
[0188][0189]
2.top1介导的dna解螺旋实验
[0190]
2.1实验材料:
[0191]
小牛胸腺dna拓扑异构酶ⅰ、负超螺旋dna质粒pbr322、琼脂糖、dmso、10x buffer缓冲液、0.1%bsa和etbr。
[0192]
2.2实验仪器
[0193]
凝胶电泳采用biorad公司powerpac电泳仪和sub-cell model 96电泳槽,凝胶扫描定量采用biorad公司的gel doc ez全自动凝胶成像系统。
[0194]
2.3实验方法
[0195]
先将1x tae溶液配置成浓度为0.8%的琼脂糖凝胶。依次向1.5ml样品管中加入10ml水,2ml buffer,2ml 0.1%bsa,top10.5u,dna 0.5ml,不同的药物0.2ml,定容到20ml。然后将样品管放入37℃水浴中,孵化15分钟。加入2ml 6x loading buffer至样品管中。110v电泳40-50分钟,用0.5mg/ml etbr染色15分钟,凝胶成像系统观察电泳结果。
[0196]
实验结果表明(如图1所示,图1是吴茱萸碱衍生物对top1和top2抑制实验结果示
意图,其中,a是吴茱萸碱衍生物在浓度为200μm时对top1的抑制实验结果示意图,b是吴茱萸碱衍生物在浓度为100μm时对top1的抑制实验结果示意图,c是吴茱萸碱衍生物在浓度为50μm时对top1的抑制实验结果示意图,条带1,超螺旋质粒dnapbr322;条带2,dna top1;条带3,dna top1 cpt;条带4-20,dna top1 目标化合物(23a,23b,23c,16a,17a,18a,16b,17b,18b,16c,17c,18c,24a,24b,24c,29a,29b)/(37d,38d,36d,37c,38c,36c,37b,38b,36b,37a,38a,36a,45a,45b,45c,29c);),所有化合物在浓度分别为200μm和100μm时均表现出top1抑制活性。它们中的大多数表现出与cpt相当或更高的top1抑制活性。在50μm的较低浓度下,五种化合物(29b-c、37d和45b-c)仍然具有活性。
[0197]
3.top2介导的dna解螺旋实验
[0198]
3.1实验材料:
[0199]
小牛胸腺dna拓扑异构酶ⅱ、负超螺旋dna质粒pbr322、琼脂糖、dmso、10x buffer缓冲液、0.1%bsa和etbr。
[0200]
3.2实验仪器
[0201]
凝胶电泳采用biorad公司powerpac电泳仪和sub-cell model 96电泳槽,凝胶扫描定量采用biorad公司的gel doc ez全自动凝胶成像系统。
[0202]
3.3实验方法
[0203]
先将1x tae溶液配置成浓度为0.8%的琼脂糖凝胶。依次向1.5ml样品管中加入10ml水,2ml buffer,2ml 0.1%bsa,top10.5u,dna 0.5ml,不同的药物0.2ml,定容到20ml。然后将样品管放入37℃水浴中,孵化15分钟。加入2ml 6x loading buffer至样品管中。110v电泳40-50分钟,用0.5mg/ml etbr染色15分钟,凝胶成像系统观察电泳结果。
[0204]
实验结果表明(如图1所示,图1中,d是吴茱萸碱衍生物在浓度为300μm时对top2的抑制实验结果示意图,e是吴茱萸碱衍生物在浓度为200μm时对top2的抑制实验结果示意图,f是吴茱萸碱衍生物在浓度为100μm时对top2的抑制实验结果示意图,条带1,超螺旋质粒dna pbr322;条带2,dna top1;条带3,dna top1 eto;条带4-16,dna top1 目标化合物(23a,23b,23c,29a,29b,29c,37a,37b,37c,37d,45a,45b,45c);g是eto和吴茱萸碱衍生物29b以及45b分别在浓度为300μm、200μm、100μm以及50μm时对top2的抑制实验结果示意图。),大多数受试化合物在300μm浓度下均表现出top2抑制活性。其中,当浓度降低到200μm和100μm时,化合物29b和45b仍然表现出显著的top2抑制活性。
[0205]
4.目标化合物体外抗肿瘤活性测试
[0206]
4.1样品配制
[0207]
用dmso(merck)溶解后,加入pbs(-)配成1000μm的溶液或均匀的混悬液,然后用含dmso的pbs(-)稀释。
[0208]
4.2细胞株
[0209]
hct116(人肠癌细胞)、mcf-7(人乳腺癌细胞)、a549(人肺癌细胞),均由本实验室冻存和传代。
[0210]
4.3培养液
[0211]
dmem或prmi1640 10%fbs 双抗
[0212]
4.4试验方法
[0213]
cck-8法:96孔板每孔加入浓度为6-10
×
104个/ml的细胞悬液100μl,置37℃,5%
co2培养箱内。24小时后,加入样品液,10ml/孔,设三复孔,37℃,5%co2作用48小时。每孔加入10mlcck-8溶液,然后37℃下避光孵育1-4小时后,用全波长多功能酶标仪测450nm od值
[0214]
实验结果显示,这些多靶点化合物具有光谱抗肿瘤活性,其中大部分化合物ic
50
值在0.18-50μm之间,其中化合物29b和45b对hct116细胞均表现出优于阳性药saha的抑制活性。
[0215]
表3.目标化合物体外抗肿瘤活性
[0216]
[0217][0218]
实施例35
[0219]
目标化合物体内抗肿瘤效果
[0220]
根据体外抗肿瘤实验结果及化合物的结构特点,选择人结肠癌hct116作为裸鼠移植瘤模型,以化合物29b和45b作为研究对象,saha作为阳性对照药。
[0221]
给药剂量设置化合物29b为20mg/kg,一天一次;化合物45b为20mg/kg或10mg/kg,一天一次;阳性药saha为20mg/kg,一天一次;连续腹腔注射给药(i.p.)14天。治疗过程中每2天监测肿瘤体积变化,肿瘤体积计算公式:(宽度2×
长度)/2。
[0222]
结果如图2所示,图2是吴茱萸碱衍生物体内抗肿瘤效果的结果示意图,其中,a是时间与肿瘤体积的结果示意图,b是肿瘤重量的实验结果示意图,c是给药结束后肿瘤照片结果示意图,经29b和45b给药处理后的肿瘤与空白组相比外形相差较大。d是时间与重量的
实验结果示意图,结果显示小鼠体重未发生明显变化,说明化合物29b和45b的体内毒性较低。
[0223]
结果显示(图2a,表4),化合物45b在剂量20mg/kg剂量下抑瘤率为69.63%;在剂量10mg/kg下,抑瘤率为55.53%,呈现剂量依赖性;化合物29b在20mg/kg剂量下抑瘤率为59.02%。结果(表4)均显著优于相同剂量下的阳性对照组(saha抑瘤率为46.01%)。此外,在给药期间并未发现小鼠体重明显变化(p》0.05,图2d),说明化合物29b和45b的体内毒性较低。
[0224]
表4.目标化合物对人体肠癌hct116裸鼠移植瘤的疗效
[0225][0226]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
