1.本发明涉及化合物的制备,尤其涉及用作医药中间体的氨基保护的苯并环酮类化合物的制备方法。例如,制备喜树碱类衍生物exatecan中用作中间体的乙酰氨基四氢萘酮类化合物。
背景技术:
2.下式ix所示的化合物为exatecan(dx-8951,一种抗肿瘤剂,参见日本专利申请公开87746/1994号),是拓扑异构酶i抑制剂喜树碱的合成类似物,在临床上用于治疗晚期软组织肉瘤,胰腺癌,食道癌,胃癌,肝癌等。其中式x化合物是exatecan全合成的重要中间体。
[0003][0004]
1991年,专利jp 3-015812中公开了以2-氟-甲苯为原料,经过七步反应得到式x化合物。此路线工艺过程安全隐患大(需要低温硼烷还原,多聚磷酸高温反应)、收率低(多聚磷酸成环一步收率只有25%)、分离困难(硝化一步有异构体副产物),不适用于工业生产。
[0005][0006]
1996年,专利wo96/26181中公开了一种新的合成路线,以2-氟-甲苯为原料,经11步合成了式x化合物。该合成方法中存在一些下述缺陷:反应过程中有多次傅克酰基化反应;羰基的还原必需经多步步骤(包括形成醇,脱水以及还原双键等);反应步骤较长,总收率只有17%。
[0007][0008]
2019年,wo2019/044946公开了日本第一三共公司研发的新合成路线,该路线以2-氟-4-硝基-甲苯为原料,经卤代,还原,氨基乙酰化,heck偶联,还原,成环共8步反应得到式x化合物。该路线引入侧链选择了heck偶联反应来完成,缩短了反应步骤,但在硝化、卤代时
收率较低,路线总收率只有14%。
[0009][0010]
由上述公开的专利可以发现氨基四氢萘酮类化合物的合成一直存在着一些问题,例如合成路线麻烦、总路线收率低等。因此,需要研制开发工业上优越的制备方法。
技术实现要素:
[0011]
本发明的目的是提供一种能高产率地制备氨基保护的苯并环酮类化合物的简便方法,其中所述化合物可用作工业制备喜树碱类衍生物(exatecan)的合成中间体。
[0012]
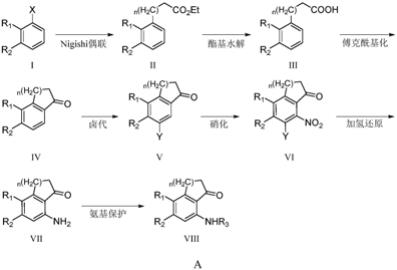
一种氨基保护的苯并环酮类化合物的制备方法,该类化合物具有式viii结构:
[0013][0014]
其中r1代表c
1-6
烷基,r2代表f或cl原子,r3代表氨基保护基,x代表br或i原子,y代表卤原子,n代表整数1~4;
[0015]
其特点在于,制备过程如下式所示,包括a,b,c,d,e,f,g共七步以下具体步骤:
[0016][0017]
a)式i化合物进行nigishi偶联制备式ii化合物;
[0018]
b)式ii化合物进行酯基水解制备式iii化合物;
[0019]
c)式iii化合物发生分子内傅克酰基化反应成环得到式iv化合物;
[0020]
d)式iv化合物在苯环上发生单卤代反应制备式v化合物;
[0021]
e)式v化合物进行硝化反应制备式vi化合物;
[0022]
f)式vi化合物加氢还原得到式vii化合物;
[0023]
g)式vii化合物进行氨基保护制备式viii化合物;
[0024]
其中:
[0025]
所述步骤a具体为:化合物i与有机锌试剂(例如4-乙氧基-4-氧代丁基溴化锌等)进行nigishi偶联反应,反应所采用溶剂优先选择无水thf或无水dmf,催化剂使用常见钯催化剂即可(三苯基膦钯、醋酸钯等),配体优先选择s-phos或x-phos,反应温度最好控制在20~60℃,反应时间最好为24~36h,锌试剂的用量优先为1.2~1.5当量,催化剂和配体的用量控制在反应物的2%mol~10%mol;
[0026][0027]
所述步骤b具体为:化合物ii溶于溶剂(低级醇、四氢呋喃等与水互溶的溶剂)中,加入碱搅拌反应。该反应选择常见的碱催化水解(例如氢氧化钠,氢氧化钾等),反应温度最好控制在0~30℃,反应时间优选1~3h,碱的用量最好为1.2~1.6当量;
[0028][0029]
所述步骤c具体为:化合物iii在酸催化下发生分子内傅克酰基化反应,使用的催化剂优选质子酸(例如硫酸、三氟乙酸或多聚磷酸等),反应最好加入脱水剂(例如五氧化二磷、三氟乙酸酐等)来促进反应进行,反应温度最好控制在-10~5℃,反应时间优选2~4h,脱水剂的用量最好为2~2.5当量;
[0030][0031]
所述步骤d具体为:化合物iv溶在硫酸或无水二氯中,分批加入溴代试剂(溴代丁二酰亚胺、二溴海因等)进行反应,反应温度最好控制在18~35℃,反应时间优选1.5~3h,溴代试剂的用量最好为1.05~1.25当量;
[0032][0033]
所述步骤e具体为:化合物v溶于溶剂质量分数为70-98%的硫酸中,低温下加入硝化试剂搅拌反应,硝化试剂选择常见的硝酸钾或硝酸即可,反应温度最好控制在-10~30℃,反应时间优选4~8h,化合物i与硝化试剂的摩尔比为1:2~2.5;
[0034][0035]
所述步骤f具体为:化合物vi在氢气氛围中加氢还原,所用溶剂优选甲醇或四氢呋喃,催化剂优选钯碳、铂碳或兰尼镍等贵金属,反应最好加入碱(例如三乙胺、吡啶等易于和卤化氢成盐的碱)来中和反应产生的酸,反应温度最好控制在15~35℃,反应时间优选18~24h,碱的用量最好为2.5~3.5当量;
[0036][0037]
所述步骤g具体为:化合物vii和氨基保护试剂反应,氨基保护试剂选用常规试剂即可(例如乙酰化试剂,乙酰化时溶剂可选用二氯甲烷、四氢呋喃等常用的无水溶剂,反应温度最好控制在15~35℃,反应时间优选1.5~2.5h,化合物vii与乙酰化试剂的摩尔比为1:2.0~3.0)。
[0038][0039]
本发明在制备氨基保护的苯并环酮类化合物时,有以下优点:首先,侧链的引入通过nigishi偶联反应来实现,缩短了反应步骤,与原有的方法相比避免了傅克酰基化、羰基还原等多步反应;其次,该合成路线发生分子内傅克酰基化成环反应时,避免了苯环上取代基的影响;最后,氨基的引入通过先引入一个溴原子来定位,与wo2019/044946公开的日本第一三共公司研发的合成路线反应相比,该方法定位效应准确,原子利用率高,经七步合成路线总收率达40%以上。
[0040]
通过本发明可以制备氨基保护的苯并环酮类化合物,该合成路线原料易得、反应类型简单、后处理方便、路线短、收率高,是适合工业制备的合成方法。
具体实施方式
[0041]
本发明所述的氨基保护的苯并环酮类化合物的制备方法在如下实施例中有更详细的叙述,但如下实施例不构成对本发明的限制。在下述实施例中,r1=甲基,r2=氟原子,r3=乙酰基,x=溴原子,y=溴原子,n=2。
[0042][0043]
实施例1制备化合物2
[0044]
在一个体积为3l的反应瓶中加入无水dmf(1000ml),2-溴-6-氟甲苯(100.0g,1.00eq),醋酸钯(2.3g,0.02eq),室温下搅拌10min后加入s-phos(8.7g,0.04eq),室温下搅拌10min后加入4-乙氧基-4-氧代丁基溴化锌(800ml,1.50eq)(注意加入锌试剂时会放出大量热!),加完后升温至30℃反应28小时。gc-ms检测原料反应完全后,将反应液冷却至室温,加入氯化铵溶液(50ml)淬灭反应后倒入10l水中,用乙酸乙酯(800ml*3)萃取,有机相合并,水洗涤两遍,饱和氯化钠溶液洗涤一遍,无水硫酸钠干燥,减压浓缩,得红棕色油状液体135.0g,粗产品不用进一步纯化,直接用于下一步反应。
[0045]
实施例2制备化合物3
[0046]
将实施例1中得到的红棕色液体(135.0g)溶于400ml甲醇中,冰水浴下滴加4m的氢氧化钠溶液(200ml,1.50eq),加完后室温反应16小时。tlc检测原料反应完全后,减压浓缩蒸去甲醇,用浓盐酸(大约70~75ml)调至ph为2,析出大量固体,过滤,干燥,得化合物3为淡黄色固体93.0g,收率90%。熔点:84.8-85.6℃。
[0047]
1h nmr(400mhz,dmso)δ12.11(s,1h),7.14(dd,j=14.2,7.1hz,1h),6.97(t,j=7.5hz,2h),2.61(t,j=7.7hz,2h),2.27(t,j=7.0hz,2h),2.17(s,3h),1.82
–
1.64(m,2h)。
[0048]
实施例3制备化合物4
[0049]
在一个1l三口瓶中加入三氟乙酸(250ml),降温至0℃分批加入化合物3(125.0g,1.00eq),再缓慢滴加三氟乙酸酐(176ml,2.00eq),加完后在该温度继续反应2.5h。tlc检测原料反应完全后,将反应液缓慢滴入冰水混合液中,再用25%w的氢氧化钠溶液调ph至7,析出大量固体,过滤,将所得固体用乙酸乙酯溶解,依次用水和饱和氯化钠溶液洗涤,无水硫酸钠干燥后,过滤,减压浓缩,得化合物4为淡黄色固体106.0g,收率93.4%。
[0050]
1h nmr(400mhz,cdcl3)δ7.95(t,j=7.1hz,1h),6.98(t,j=8.8hz,1h),2.88(t,j=5.8hz,2h),2.69
–
2.56(m,2h),2.22(s,3h),2.16(dt,j=11.9,6.1hz,2h)。
[0051]
实施例4制备化合物5
[0052]
在2l的三口瓶中加入浓硫酸(1l),冰水浴下分四批加入化合物4(100.0g,1.00eq),完全溶解后分五批加入nbs(110.0g,1.10eq),加完后升至室温反应2小时。tlc检测反应完全后,将反应液缓慢滴入10l冰水混合液中,析出固体,搅拌两小时,过滤。固体用乙酸乙酯溶解,依次用饱和碳酸钠溶液,水,饱和氯化钠溶液洗涤,有机相用无水硫酸钠干燥,减压浓缩,得化合物5为淡黄色固体115g,收率80%。
[0053]
1h nmr(400mhz,dmso)δ7.95(d,j=7.0hz,1h),2.90
–
2.79(m,2h),2.59(t,j=
10.5hz,2h),2.24(s,3h),2.12
–
1.98(m,2h)。
[0054]
实施例5制备化合物6
[0055]
在500ml的三口瓶中加入浓硫酸(300ml),冰水浴下加入化合物5(30.00g,1.00eq),待完全溶解后降温至0℃,缓慢滴加硝酸(19.30ml,2.50eq),滴加完后维持该温度搅拌反应。tlc检测原料反应完全后,将反应液缓慢倒入冰水混合液中,析出固体,过滤。干燥,用pe:ea=10:1的混合溶液搅拌2小时,过滤,干燥,得化合物6为亮黄色固体28g,收率81%。
[0056]
1h nmr(400mhz,dmso)δ2.93(d,j=4.1hz,2h),2.66(d,j=4.6hz,2h),2.30(s,3h),2.09(d,j=5.0hz,2h)。
[0057]
实施例6制备化合物7
[0058]
在3l的瓶中加入化合物6(50.00g,1.00eq)、甲醇(1.5l)、三乙胺(68.8ml,3.00eq)和钯碳(2.50g,5wt%),氢气氛围中室温反应,反应24小时。tlc检测原料反应完全后,将反应液过滤,滤液减压浓缩,真空泵抽干,直接用于下一步反应。
[0059]
实施例7制备化合物8
[0060]
将实施例6中所得产品置于1l圆底烧瓶中,加入无水dcm(320ml)和吡啶(46.75ml,3.50eq),缓慢滴加乙酰氯(3.00eq,35.45ml),加完后室温下反应2h,tlc检测原料反应完全后,将反应液依次用稀盐酸,碳酸氢钠溶液、水、饱和氯化钠洗涤,有机相用无水硫酸钠干燥,减压浓缩,得化合物8为淡红色固体31g,收率80%。
[0061]
1h nmr(400mhz,cdcl3)δ12.31(s,1h),8.40(d,j=12.9hz,1h),2.88(t,j=6.1hz,2h),2.66(t,j=6.4hz,2h),2.22(s,3h),2.14(d,j=1.4hz,3h),2.12
–
2.05(m,2h)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
