1.本发明公开了一种质粒,属于核酸分子技术领域。
背景技术:
2.抗体(antibody,ab)是介导体液免疫的重要效应分子,是b细胞接受抗原刺激后增值分化为浆细胞后所产生的糖蛋白,主要存在于血清等体液中,能与相应抗原特异性结合,进而来发挥免疫功能,适应症覆盖自身免疫性疾病、实体瘤和血液肿瘤等领域。
3.单克隆抗体药物具有以下趋势:(1)抗体治疗领域得到极大扩展,从传染病到肿瘤(淋巴瘤、胃癌、肺癌等)、自身免疫疾病(银屑病、系统性红斑狼疮等)、代谢性疾病(高脂血症等)或者退行性病变(黄斑变性、阿尔兹海默症)。(2)整个研发生产销售链条非常完善,从基因发现,抗体上游(载体构建、克隆表达)到下游(规模化发酵和纯化质量控制、杂质去除)以及产业化(生产、全球销售、部分产品进入医保)。(3)新技术层出不穷,从单克隆抗体技术到双特异抗体、多特异抗体,抗体药物偶联物,纳米抗体多种抗体药物发展。(4)给药方式多样:从经典的静脉和腹腔给药到皮下注射,眼内注射,鼻腔吸入等。
4.抗传染病抗体药物伴随抗体药物的发展而进入高速发展期,从呼吸道合胞病毒治疗性抗体到炭疽治疗性抗体药物,再到最近的埃博拉抗体治疗药物和covid-19治疗性抗体药物。从某种程度上讲,抗体药物因为靶标明确,作用机理相对清晰,成为各大制药公司的战略布局方向。covid-19治疗性抗体药物研发中,再生元、礼来、阿斯利康、vir等制药巨头研发进度居领先地位。
5.传染病抗体药物的发现从杂交瘤技术,到噬菌体文库技术,到转基因鼠技术再到现在的单细胞pcr技术、单细胞测序技术。相比以前的发现技术,单细胞技术具有明显技术进步,基因来源于康复者或者免疫志愿者的外周血单个b淋巴细胞,为天然配对的全人源抗体基因,基本避免以前抗体研发的免疫原性和稳定性不足的缺陷。
6.无论哪种发现策略,最后需要将轻链和重链的全长基因克隆到真核表达载体上,制备全分子的抗体,然后进行体内、外活性评价,才能进一步评估是否具有成药性。目前,相对优选的策略是来源于外周血的全分子抗体候选分子直接进行筛选和评价。常规的方法是构建线性表达框的策略进行结合活性筛选,缺点是表达量少,常常存在突变,有时序列不唯一。筛选获得具有结合活性的抗体基因后,需要将抗体基因克隆到t载体上进行测序确认后,再通过酶切亚克隆到表达载体。有的商品化载体可以同时实现克隆和表达载体构建,缺点是存在个别位点的氨基酸突变,这样使得药物研发中还得进行氨基酸的校正突变,增加了复杂性。
技术实现要素:
7.本发明的目的是提供一种可以便捷、快速构建人源igg1/kappa、igg1/lambda抗体表达载体的方法,可以将抗体轻重链的可变区基因通过同源重组的方式快速地克隆至包含有信号肽和编码抗体恒定区基因的质粒中,即可进行抗体的真核表达。
8.基于上述目的,本发明首先提供了一种人源抗体表达质粒系统,所述人源抗体表达质粒系统包括人源抗体重链表达质粒和人源抗体轻链表达质粒,在所述人源抗体重链表达质粒含有重链可变区同源臂重组序列,所述人源抗体重链编码基因通过自身携带的,并与重链可变区同源臂重组序列相同的同源臂重组序列通过同源重组整合到所述人源抗体重链表达质粒中,以及,在所述人源抗体轻链表达质粒含有轻链可变区同源臂重组序列,所述人源抗体轻链编码基因通过自身携带的,并与轻链可变区同源臂重组序列相同的同源臂重组序列通过同源重组整合到所述人源抗体轻链表达质粒中。
9.在一个优选的实施方案中,所述重链可变区上游同源臂重组序列如seqidno.12所示,所述轻链可变区上游同源臂重组序列如seqidno.14或seqidno.16所示。
10.在一个更为优选的实施方案中,所述人源抗体重链表达质粒还含有抗体重链信号肽编码基因和抗体重链恒定区编码基因,以及,所述人源抗体请链表达质粒还含有抗体轻链信号肽编码基因和抗体轻链恒定区编码基因。
11.更为优选地,在编码抗体重链信号肽的下游基因和编码抗体重链恒定区的基因上游分别引入hpai和srfi两个酶切位点。
12.尤为优选地,所述抗体重链信号肽编码基因和抗体重链恒定区编码基因之间的序列如seqidno.1所示。
13.在另一个优选的实施方案中,所述抗体轻链为kappa链,在编码kappa链信号肽的下游基因和编码恒定区的基因上游分别引入kpni和pmei两个酶切位点。
14.更为优选地,在所述kappa链信号肽编码基因和kappa链恒定区编码基因之间的序列如seqidno.2所示。
15.在又一个优选的实施方案中,所述抗体轻链为lambda链,在编码lambda链信号肽的下游基因和编码恒定区的基因上游分别引入kpni和pmli两个酶切位点。
16.更为优选地,在所述lambda链信号肽编码基因和lambda链恒定区编码基因之间的序列如seqidno.3所示。
17.在本发明的一个优选的实施方案中,所述抗体重链信号肽编码基因的序列如seqidno.18所示,抗体重链恒定区编码基因的序列如seqidno.19(genbank:aaa02914.1)所示。
18.在本发明的另一个优选的实施方案中,所述抗体kappa链信号肽编码基因的序列如seqidno.20所示,抗体kappa链恒定区编码基因的序列如seqidno.21(genbank:aay24201.1)所示。
19.在本发明的又一个优选的实施方案中,所述抗体lambda链信号肽编码基因的序列如seqidno.22所示,抗体lambda链恒定区编码基因的序列如seqidno.23(genbank:aah12159.1)所示。
20.其次,本发明还提供了上述的人源抗体表达质粒系统的制备方法,所述方法包括以下步骤:(1)获得具有抗体重链信号肽编码基因和抗体重链恒定区编码基因的基因片段,在抗体重链信号肽编码基因和抗体重链恒定区编码基因引入限制性酶切位点以及非抗体基因相关基因;或者获得具有抗体kappa轻链信号肽编码基因和抗体kappa轻链恒定区编码基因的基
因片段,在抗体kappa轻链信号肽编码基因和抗体kappa轻链恒定区编码基因引入限制性酶切位点以及非抗体基因相关基因;或者获得具有抗体lambda轻链信号肽编码基因和抗体lambda轻链恒定区编码基因的基因片段,在抗体lambda轻链信号肽编码基因和抗体lambda轻链恒定区编码基因引入限制性酶切位点以及非抗体基因相关基因;(2)获得含有重链可变区同源臂重组序列,以及具有人巨细胞病毒早期启动子元件cmv、增强表达原件wpre、poly a 尾基因、氨苄抗性基因的真核细胞表达载体的双酶切片段;(3)将步骤1获得基因片段利用同源重组方法克隆至步骤(2)表达载体的双酶切片段。
21.最后,本发明还提供了一种应用上述的人源抗体表达质粒系统制备人源化抗体的方法,所述方法包括以下步骤:(1)使线性化的人源抗体重链表达质粒和人源抗体轻链表达质粒分别与待制备抗体的重链可变区基因与轻链可变区基因进行同源重组,其中,在重链可变区基因与轻链可变区基因的5’端分别设置有与重链可变区同源臂重组序列和轻链可变区同源臂重组序列相同的同源臂重组序列;(2)表达步骤(1)获得的携带有重链可变区基因与轻链可变区基因的人源抗体重链表达质粒和人源抗体轻链表达质粒,并回收表达产物。
22.在一个优选的实施方案中,所述重链可变区同源臂重组序列如seq id no.12所示,所述轻链可变区同源臂重组序列如seq id no.14或seq id no.16所示。
23.相比传统的方法,本发明提供的方法具有下面的进步:1省时:信号肽和恒定区直接构建在载体上,不需要进行融合pcr获得全长抗体基因过程,有效避免融合pcr过程导致的序列突变问题。
24.2高通量:构建的载体统一酶切后回收,通过单细胞pcr技术或者单细胞测序技术获得的可变区pcr产物直接通过96孔板进行回收,通过同源重组连接到载体上。
25.3简便:同源重组后的菌落无需进行克隆pcr鉴定,直接选取菌落进行序列测定确认,正确率高大于90%。不需要构建表达框可直接进行表达载体构建。
附图说明
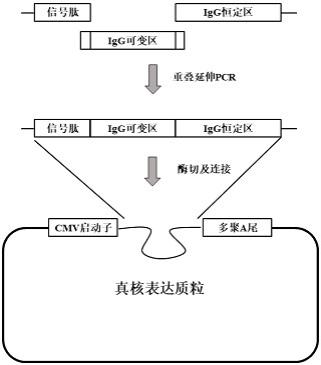
26.图1为实施例抗体常规表达载体构建流程示意图;图2为实施例pabh表达载体构建流程示意图;图3为实施例pabk表达载体构建流程示意图;图4为实施例pabλ表达载体构建流程示意图;图5为实施例设计基因的pcr扩增结果示意图;图6为实施例pab载体的双酶切示意图;图7为抗体e2和e40轻重链可变区电泳图;图8为实施例抗体e2和e40轻重链表达载体双酶切电泳图;图9为实施例抗体纯化后sds-page电泳图;图10为实施例抗体elisa分析图。
具体实施方式
27.下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的权利要求所限定的保护范围构成任何限制。
28.本发明以针对裂谷热病毒gn蛋白的抗体e2和e40的表达质粒的构建过程为例进行说明,其中e2是一株轻链为kappa型的抗体,e40是一株轻链为lambda型的抗体。
29.实施例1:针对裂谷热病毒gn蛋白的抗体e2和e40的表达质粒的构建1.实施例中所涉及试剂:hpa i、srf i、kpn i、pme i和pml i限制性内切酶和同源重组酶nebuilder购自neb公司。胶回收试剂盒购自omega公司。hitrap rproteina购自ge公司。hrp标记的羊抗人igg二抗购自abcam公司。tanstart taqdna聚合酶购自北京全式金公司。expi293f细胞培养基及转染试剂购自thermo fisher公司。实施例中抗体e2和e40可变区基因及所用引物和测序均由生工生物工程公司合成和完成。top10感受态购自博迈德公司。
30.2.载体设计及构建:采用合适的表达载体是成功表达外源基因的关键,作为能够进行高表达适合工业规模化应用的哺乳动物细胞表达载体必须具备:强的启动子和高效的转录终止序列;增强表达的内含子或者元件。本发明拟将设计的基因克隆到通用的真核细胞表达载体pab上,本载体的技术特征是:(1)具有人巨细胞病毒早期启动子元件(cmv),是众多哺乳动物细胞中启动表达的较强的启动子,有很广泛的宿主适用范围,其中包括中国仓鼠卵巢细胞(chinese hamster ovary,cho)。(2)增强表达的原件wpre,转录后调控序列,增加外源片段的表达效率。(3) poly a 尾基因,有助mrna从核到细胞质转运以及避免mrna在细胞中受到核酶降解,增强mrna的稳定性(4)氨苄抗性基因:在含有氨苄抗生素的lb平板上,成功转化的阳性细菌克隆能够生长。常规克隆策略见图1,包括信号肽、可变区、恒定区的融合,测序正确后克隆到载体的相应酶切位点,构建表达质粒。
31.本发明在不改变氨基酸序列(人igg1重链恒定区参考genbank: aaa02914.1、人kappa轻链恒定区参考genbank: aay24201.1,人lambda轻链恒定区参考genbank: aah12159.1,氨基酸序列的条件下,通过同义突变引入单一酶切位点的方式对3条分别表达人源抗体igg1重链、kappa链和lambda链的基因进行改造。在编码重链信号肽的下游基因(seq id no.18)和编码恒定区的基因(seq id no.19)上游分别引入hpa i(gtt^aac)和srf i(gccc^gggc)两个酶切位点,在编码kappa链信号肽的下游基因(seq id no.20)和编码恒定区的基因(seq id no.21)序列上游分别引入kpn i (ggt^acc)和pme i(gttt^aaac)两个酶切位点,在编码lambda链信号肽的下游基因(seq id no.22)和编码链恒定区的基因(seq id no.23)上游分别引入kpn i(ggt^acc)和pml i(cac^gtg)两个酶切位点,具体见附图2、图3和图4。然后,在上述三条基因中的两个酶切位点之间引入一段与抗体基因无关的序列。设计的全长基因序列分别见seq id no.1、seq id no.2、seq id no.3。最后将三条基因序列送交生物公司合成。
32.2.1全基因化学合成根据设计的序列委托安徽通用生物技术公司全基因化学合成。
33.2.2含轻重链恒定区基因的全长扩增
通过上下游引物abhr-f和abhr-r,利用neb2
×
q5dna聚合酶对seqidno.1、seqidno.2、seqidno.3基因扩增。引物序列见表1:。
34.备注:gaattc为ecori酶切位点;ggatcc为bamhi酶切位点;ccttggatctctagc为上游同源臂;cttgtcgaggtcggg为下游同源臂。反应体系见表2: 表2.基因扩增反应体系。
35.反应程序见表3:表3.基因扩增反应程序。
36.扩增结果见图5,泳道1、2、3分别是重链、kappa链、lambda链扩增结果,条带大小与预期相符。
37.2.3质粒pab的双酶切线性化及回收纯化使用限制性内切酶ecori和bamhi对质粒pab进行双酶切,使其线性化。酶切体系如表4:表4.质粒pab双酶切反应体系
。
38.酶切结果见图6,泳道1显示载体被线性化。
39.2.4连接,转化及阳性克隆筛选:目的片段与载体的连接是使用nebuilder组装试剂盒 (nebuilder
™ꢀ
高效无缝克隆技术是neb自主研发的一项高效同源重组技术,它利用目的片段两端和载体两端含有相同的序列,在nebuilder
™ꢀ
master mix作用下完成高效的同源重组,将信号肽和恒定区基因先克隆到载体上,构建重组载体),按说明书进行操作,连接目的片段。反应体系如表5:表5. 同源重组反应体系。
40.然后轻微混匀,50 ℃连接15 min,随后将体系置于冰上。取5 μl连接产物加入top10感受态中,冰浴30 min,42 ℃热激90 s,冰浴3 min 后,使用无抗性lb培养基,振荡培养60分钟,8000 rpm离心1分钟,取沉淀重悬液100 μl,涂板,37℃温箱孵育过夜。用无菌的枪头挑取生长状态良好的单克隆菌落到装有600 μl amp/lb液体培养基的1.5 ml无菌离心管中,置于37 ℃摇床培养12 h,随机挑选抗性克隆进行测序验证。
41.获得的载体命名为pabh、pabk和pabλ,分别含有igg1重链恒定区、kappa链恒定区和λ链恒定区,相应的序列分别见seq id no.1、seq id no.2和seq id no.3,相应的序列功能分别在表6、表8和表10中进行注释。
42.表6.ab-h序列(seq id no.1)注释
ꢀ
。
43.重链上游同源臂核苷酸序列:ctaattttaaaaggtgtt(seqidno.12),长度18bp,分别位于载体上和可变区上游引物上,负责同源重组。
44.igg1重链恒定区5端同源臂:ccagggggaagaccgatgggccc(seqidno.13),25bp,分别位于载体上和可变区下游引物上,负责同源重组。
45.表7.扩增重链可变区的引物序列 。
46.表8.ab-k序列注释(seqidno.2)
。
47.kappa链上游同源臂核苷酸序列:ctgctatgggtatctggt(seq id no.14),18bp,分别位于载体上和可变区上游引物上,负责同源重组。
48.kappa链恒定区5端同源臂:gaagacagatggtgcagccacagtacgttt(seq id no.15), 30bp,分别位于载体上和可变区下游引物上,负责同源重组。
49.表9. 扩增kappa链可变区的引物序列。
50.表10. ab-l序列注释(seq id no.3)
。
[0051] lambda链上游同源臂核苷酸序列:ctgctatgggtatctggt(seq id no.16),18bp,分别位于载体上和可变区上游引物上,负责同源重组lambda链恒定区5端同源臂:gggggcagccttgggctgccc(seq id no.17), 21bp,分别位于载体上和可变区下游引物上,负责同源重组表11. 扩增lambda链可变区的引物序列。
[0052]
实施例2.抗体制备实施例1 单细胞pcr扩增抗体可变区基因首先,通过表2中的e2和e40轻重链可变区引物,利用transtart taqdna聚合酶对抗体的可变区基因进行pcr扩增,相应的引物序列见表3、表5和表7。反应体系如表12:表12. 基因扩增反应体系
。
[0053]
反应程序如表13:表13.基因扩增反应程序。
[0054]
实施例抗体轻重链可变区基因均得到了很好的扩增,大小均符合预期大小(约400bp),结果见附图7(左为e2重链(seqidno.24序列)和kappa链可变区基因(seqidno.25序列)的扩增;右为e40e2重链(seqidno.26,序列)和kappa链可变区基因(seqidno..27,序列)的扩增),分子大小在400bp,符合预期。
[0055]
2质粒pabh、pabκ和pabλ的双酶切线性化及回收纯化使用相应的限制性内切酶对质粒pabh、pabκ和pabλ进行双酶切,使其线性化。酶切体系如表14-表16:表14.pabh双酶切反应体系
ꢀ
。
[0056]
表15.pabκ双酶切反应体系 表16.pabλ双酶切反应体系。
[0057]
37℃酶切1h。然后按照omegagelextractionkit进行胶回收纯化。过程如下:(1)切胶及称量:将含有目的基因的琼脂糖凝胶切下,并尽可能切除多余部分,保
留目的条带。将切下凝胶块至于干净的1.5 ml离心管中,并称取凝胶块的重量,向胶块中加入等倍体积的binding buffer(凝胶块质量为1 mg,其体积可视为1 μl,则加入1μl binding buffer溶液),置于65℃金属浴中直至凝胶块完全溶解。
[0058]
(2)目的条带与吸附柱结合:将溶解充分于binding buffer的凝胶块加入吸附柱中,12000rpm 离心1 min,弃掉收集管中的废液,并将吸附柱中重新置于收集管中。再取300μl binding buffer 加入到吸附柱中,12000 rpm离心1 min,弃掉收集管中的废液。
[0059]
(3)洗涤:向上述已结合dna片段的吸附柱中加入700 μl wash buffer(使用前确认加入无水乙醇),12000 rpm离心1min,弃掉收集管中的废液,将吸附柱重新置于收集管中,此步骤重复两次。将吸附柱重新置于收集管中,12000 rpm空离2 min,并在室温中干燥5-10 min使wash buffer中酒精充分蒸发,以免影响下一步实验。
[0060]
(4)目的片段洗脱:将上述吸附柱置于一个干净的1.5 ml离心管中,向吸附膜中央滴加40 μl ddh2o 置于65 ℃金属浴中作用5min,使dna充分溶解于ddh2o。最后,12000rpm离心2 min收集dna溶液。
[0061]
3连接,转化及阳性克隆筛选:目的片段与载体的连接是使用nebuilder组装试剂盒,并按照说明书进行操作((nebuilder
™ꢀ
高效无缝克隆技术是neb自主研发的一项高效同源重组技术,它利用目的片段两端和载体两端含有相同的序列,在nebuilder
™ꢀ
master mix作用下完成高效的同源重组,构建重组载体)。按说明书进行操作,连接目的片段。反应体系如表17.表17. 同源重组反应体系。
[0062]
然后轻微混匀,50 ℃连接15 min,随后将体系置于冰上。取5 μl连接产物加入top10感受态中,冰浴30 min,42 ℃热激90 s,冰浴3 min 后,使用无抗性lb培养基,振荡培养60分钟,8000 rpm离心1分钟,取沉淀重悬液100 μl,涂板,37℃温箱孵育过夜。用无菌的枪头挑取生长状态良好的单克隆菌落到装有600 μl amp/lb液体培养基的1.5 ml无菌离心管中,置于37 ℃摇床培养12 h,随机挑选抗性克隆进行测序验证。
[0063]
结果:e2单抗重链随机挑选抗性克隆4个送测序,其中序列比对符合预期的有4个,阳性率为100%;e40单抗重链随机挑选抗性克隆4个送测序,其中序列比对符合预期的有3个,阳性率为75%;kappa链随机挑选抗性克隆4个送测序,其中序列比对符合预期的有4个,阳性率为100%;lambda链随机挑选抗性克隆4个送测序,其中序列比对符合预期的有4个,阳性率为100%。对e2和e40单抗的轻重链可变区合计4个片段使用本技术成功进行质粒构建,送16个克隆进行序列测定,其中15个克隆符合预期,阳性率15/16=93.75%。构建的表达载体
进行双酶切鉴定,酶切结果见附图8,其中泳道pe2-h显示e2单抗重链载体双酶切结果,预期载体条带约7000bp和重链条带约1500bp;泳道pe2-l显示e2单抗轻链载体双酶切结果,预期载体条带约7000bp和轻链条带约750bp; 其中泳道pe40-h显示e40单抗重链载体双酶切结果,预期载体条带约7000bp和重链条带约1500bp;泳道pe40-l显示e40单抗轻链载体双酶切结果,预期载体条带约7000bp和轻链条带约750bp。
[0064]
4.抗体的真核表达与纯化将上述测序正确的阳性抗体轻重链表达质粒进行抽提。然后将配对的轻重链质粒共转染至expi293f细胞中。转染前,将总细胞数为3
×
106个expi293f 细胞接种于30 ml expi293f表达培养基中,将细胞置于摇床中,5% co2,37 ℃,120 rpm的条件下持续培养数小时,当细胞密度达3.5
×
106/ml时,将80 μl expifectamine293转染试剂加入1.5 ml培养基中混匀,室温孵育5 min。将30 μg表达质粒加入1.5 ml培养基中混匀。然后将孵育好的转染试剂与培养基的混合物加入质粒与培养基的混合物中轻轻混匀,室温孵育25 min,最后缓慢加入细胞培养瓶中。在培养16 h后,在上述细胞培养瓶中分别加入150 μl转染增强剂1 和1.5 ml转染增强剂2。
[0065]
收取连续培养108 h后的细胞上清,首先以4 ℃,1500 g离心15 min,然后再以3000 g离心15 min,将上清转移至新的离心管中,最后再以12000 rpm高速离心10 min后,将高速离心后的上清转移至新的离心管中,置于4 ℃备用。
[0066]
为了将expi293f细胞表达的抗体从细胞培养基中分离出来,我们通过hitrap rproteina亲和柱对抗体进行了纯化,步骤如下:1)首先,将hitrap rproteina亲和柱以低流速接于蛋白纯化仪akta pure中,然后走5个柱体积的纯水将rproteina亲和柱中20%的乙醇冲洗干净,再走平衡缓冲液(pbs),直至uv走平后将uv设置调零。
[0067]
2)将上述高速离心后的细胞上清以正常流速上样,并收集柱后,上样完成后,继续用pbs平衡直至uv走平。
[0068]
3)用ph 2.7的0.1 m甘氨酸洗脱亲和柱中抗体,并收集洗脱峰。并用ph 9.0的tris-hcl溶液将洗脱液中和至ph 7.0。然后通过sds-page分析纯化结果。
[0069]
4)将纯化成功的抗体通过30kda的超滤管进行浓缩换液至pbs中,离心条件为4 ℃,3000 g离心30 min。
[0070]
最后对纯化后的抗体sds-page分析,实施例抗体均得到了良好的表达及纯化,结果见附图9。该结果说明,本发明在氨基酸序列不改变的条件下,所进行的碱基突变并不影响抗体的正常表达。
[0071]
5 .elisa测定纯化后gn蛋白单克隆抗体的结合活性取纯化的截短型gn蛋白包被于elisa板(2 μg/ml,100 μl/孔)4 ℃过夜,pbst洗涤3次后,用2% bsa于37 ℃封闭1 h后;pbst洗涤3次。首孔加150 μl浓度为10 μg/ml的抗体,然后进行三倍梯度稀释,共稀释12个稀释度,然后置于37 ℃孵育1 h后,pbst洗涤三次。每孔加入100 μl hrp标记的羊抗人igg二抗(1:10000稀释),置于37 ℃孵育1 h后,pbst洗涤三次。每孔加入100 μl tmb单组份显色液,室温显色5 min,再加入50 μl终止液,最后用酶标仪读取od450nm-od630nm值。最后利用graphpad prism软件对每个单克隆抗体的结合曲线进行四参数拟合,测得e2和e40的ec
50
分别为2.157ng/ml,4.565ng/ml,结果见附图10。该
结果说明,本发明在氨基酸序列不改变的条件下,所进行的碱基突变并不影响抗体与抗原的结合功能。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
